
Localization of Epigenetic Markers in Leishmania Chromatin

 and
and
Abstract
1. Introduction
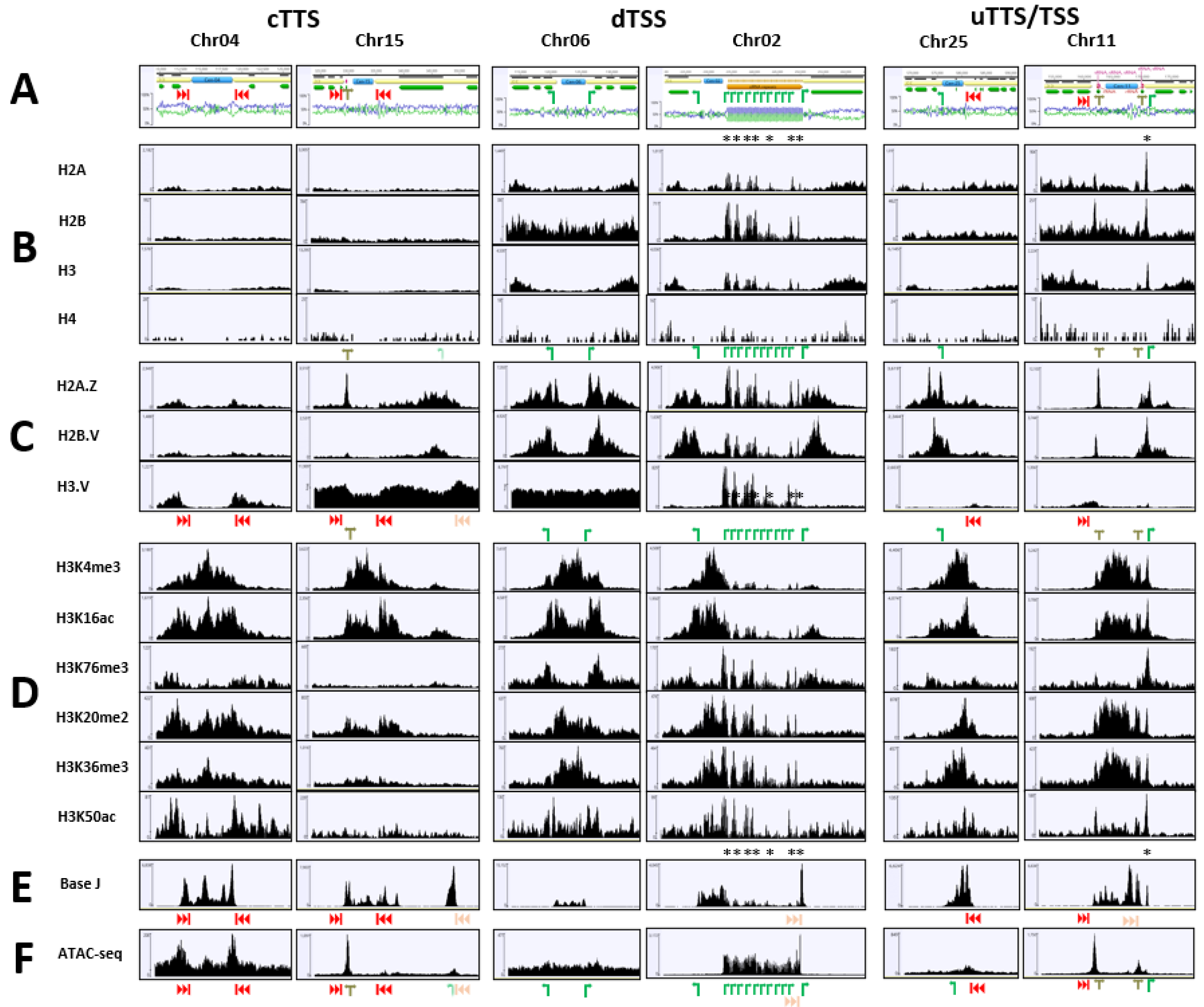
2. Results
3. Discussion
4. Materials and Methods
4.1. Parasite Cell Culture
4.2. Assembly and Annotation of the Reference Genome
4.3. CUT & Tag
4.4. J-IP-Seq
4.5. ATAC-Seq
4.6. Sequencing and Read Mapping
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCall, L.I.; McKerrow, J.H. Determinants of disease phenotype in trypanosomatid parasites. Trends Parasitol 2014, 30, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Russell, D.G. The interaction of Leishmania parasites with macrophages. Adv. Parasitol. 1992, 31, 175–254. [Google Scholar] [PubMed]
- Cunningham, A.C. Parasitic adaptive mechanisms in infection by Leishmania. Exp. Mol. Pathol. 2002, 72, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of histone modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar]
- Padilla-Mejia, N.E.; Florencio-Martinez, L.E.; Figueroa-Angulo, E.E.; Manning-Cela, R.G.; Hernandez-Rivas, R.; Myler, P.J.; Martinez-Calvillo, S. Gene organization and sequence analyses of transfer RNA genes in trypanosomatid parasites. BMC Genom. 2009, 10, 232. [Google Scholar] [CrossRef]
- Borst, P. Discontinuous transcription and antigenic variation in trypanosomes. Annu. Rev. Biochem. 1986, 55, 701–732. [Google Scholar] [CrossRef]
- Clayton, C.E. Gene expression in kinetoplastids. Curr. Opin. Microbiol. 2016, 32, 46–51. [Google Scholar] [CrossRef]
- Thomas, S.; Green, A.; Sturm, N.R.; Campbell, D.A.; Myler, P.J. Histone acetylations mark origins of polycistronic transcription in Leishmania major. BMC Genom. 2009, 10, 152. [Google Scholar] [CrossRef]
- Siegel, T.N.; Hekstra, D.R.; Kemp, L.E.; Figueiredo, L.M.; Lowell, J.E.; Fenyo, D.; Wang, X.; Dewell, S.; Cross, G.A. Four histone variants mark the boundaries of polycistronic transcription units in Trypanos brucei. Genes Dev. 2009, 23, 1063–1076. [Google Scholar] [CrossRef]
- Wright, J.R.; Siegel, T.N.; Cross, G.A. Histone H3 trimethylated at lysine 4 is enriched at probable transcription start sites in Trypanos brucei. Mol. Biochem. Parasitol. 2010, 172, 141–144. [Google Scholar] [CrossRef]
- Maree, J.P.; Patterton, H.G. The epigenome of Trypanosoma brucei: A regulatory interface to an unconventional transcriptional machine. Biochim. Biophys. Acta 2014, 1839, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Gommers-Ampt, J.H.; Van Leeuwen, F.; De Beer, A.L.J.; Vliegenthart, J.F.G.; Dizdaroglu, M.; Kowalak, J.A.; Crain, P.F.; Borst, P. β-d-glucosyl-hydroxymethyluracil: A novel modified base present in the DNA of the parasitic protozoan T. Brucei. Cell 1993, 75, 1129–1136. [Google Scholar] [CrossRef]
- Van Leeuwen, F.; Taylor, M.C.; Mondragon, A.; Moreau, H.; Gibson, W.; Kieft, R.; Borst, P. β-d-glucosyl-hydroxymethyluracil is a conserved DNA modification in kinetoplastid protozoans and is abundant in their telomeres. Proc. Natl. Acad. Sci. USA 1999, 95, 2366–2371. [Google Scholar] [CrossRef] [PubMed]
- van Luenen, H.G.; Farris, C.; Jan, S.; Genest, P.A.; Tripathi, P.; Velds, A.; Kerkhoven, R.M.; Nieuwland, M.; Haydock, A.; Ramasamy, G.; et al. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell 2012, 150, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Ekanayake, D.K.; Minning, T.; Weatherly, B.; Gunasekera, K.; Nilsson, D.; Tarleton, R.; Ochsenreiter, T.; Sabatini, R. Epigenetic regulation of transcription and virulence in Trypanosoma cruzi by o-linked thymine glucosylation of DNA. Mol. Cell. Biol. 2011, 31, 1690–1700. [Google Scholar] [CrossRef]
- Cliffe, L.J.; Siegel, T.N.; Marshall, M.; Cross, G.A.; Sabatini, R. Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanos brucei. Nucleic Acids Res. 2010, 38, 3923–3935. [Google Scholar] [CrossRef]
- Reynolds, D.; Cliffe, L.; Forstner, K.U.; Hon, C.C.; Siegel, T.N.; Sabatini, R. Regulation of transcription termination by glucosylated hydroxymethyluracil, base J, in Leishmania major and Trypanosoma brucei. Nucleic Acids Res. 2014, 42, 9717–9729. [Google Scholar] [CrossRef]
- Ekanayake, D.; Sabatini, R. Epigenetic regulation of polymerase ii transcription initiation in Trypanosoma cruzi: Modulation of nucleosome abundance, histone modification, and polymerase occupancy by o-linked thymine DNA glucosylation. Eukaryot Cell 2011, 10, 1465–1472. [Google Scholar] [CrossRef]
- Janzen, C.J.; Fernandez, J.P.; Deng, H.; Diaz, R.; Hake, S.B.; Cross, G.A. Unusual histone modifications in Trypanosoma brucei. FEBS Lett. 2006, 580, 2306–2310. [Google Scholar] [CrossRef]
- Horn, D. Introducing histone modification in trypanosomes. Trends Parasitol. 2007, 23, 239–242. [Google Scholar] [CrossRef][Green Version]
- Mandava, V.; Fernandez, J.P.; Deng, H.; Janzen, C.J.; Hake, S.B.; Cross, G.A. Histone modifications in Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 156, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Picchi, G.F.; Zulkievicz, V.; Krieger, M.A.; Zanchin, N.T.; Goldenberg, S.; de Godoy, L.M. Post-translational modifications of Trypanosoma cruzi canonical and variant histones. J. Proteome Res. 2017, 16, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Fasel, N.J.; Robyr, D.C.; Mauel, J.; Glaser, T.A. Identification of a histone H1-like gene expressed in Leishmania major. Mol. Biochem. Parasitol. 1999, 62, 321–324. [Google Scholar] [CrossRef]
- Kraus, A.J.; Vanselow, J.T.; Lamer, S.; Brink, B.G.; Schlosser, A.; Siegel, T.N. Distinct roles for H4 and H2A.Z acetylation in RNA transcription in African trypanosomes. Nat. Commun. 2020, 11, 1498. [Google Scholar] [CrossRef]
- Mandava, V.; Janzen, C.J.; Cross, G.A. Trypanosome H2B.V replaces H2B in nucleosomes enriched for H3 K4 and K76 trimethylation. Biochem. Biophys. Res. Commun. 2008, 368, 846–851. [Google Scholar] [CrossRef]
- Schulz, D.; Zaringhalam, M.; Papavasiliou, F.N.; Kim, H.S. Base J and H3.V regulate transcriptional termination in Trypanosoma brucei. PLoS Genet 2016, 12, e1005762. [Google Scholar] [CrossRef]
- Anderson, B.A.; Wong, I.L.; Baugh, L.; Ramasamy, G.; Myler, P.J.; Beverley, S.M. Kinetoplastid-specific histone variant functions are conserved in Leishmania major. Mol. Biochem. Parasitol. 2013, 191, 53–57. [Google Scholar] [CrossRef]
- Janssen, K.A.; Sidoli, S.; Garcia, B.A. Recent cchievements in characterizing the histone code and approaches to integrating epigenomics and systems biology. Methods Enzymol. 2017, 586, 359–378. [Google Scholar]
- Kumar, D.; Rajanala, K.; Minocha, N.; Saha, S. Histone H4 lysine 14 acetylation in Leishmania donovani is mediated by the myst-family protein HAT4. Microbiology 2012, 158, 328–337. [Google Scholar] [CrossRef][Green Version]
- Jha, P.K.; Khan, M.I.; Mishra, A.; Das, P.; Sinha, K.K. HAT2 mediates histone H4K4 acetylation and affects micrococcal nuclease sensitivity of chromatin in Leishmania donovani. PLoS ONE 2017, 12, e0177372. [Google Scholar] [CrossRef]
- Chandra, U.; Yadav, A.; Kumar, D.; Saha, S. Cell cycle stage-specific transcriptional activation of cyclins mediated by HAT2-dependent H4K10 acetylation of promoters in Leishmania donovani. PLoS Pathog. 2017, 13, e1006615. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Okur, H.S.; Janssens, D.H.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 2020, 15, 3264–3283. [Google Scholar]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [PubMed]
- Garcia-Silva, M.R.; Sollelis, L.; MacPherson, C.R.; Stanojcic, S.; Kuk, N.; Crobu, L.; Bringaud, F.; Bastien, P.; Pages, M.; Scherf, A.; et al. Identification of the centromeres of Leishmania major: Revealing the hidden pieces. EMBO Rep. 2017, 18, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Lombrana, R.; Alvarez, A.; Fernandez-Justel, J.M.; Almeida, R.; Poza-Carrion, C.; Gomes, F.; Calzada, A.; Requena, J.M.; Gomez, M. Transcriptionally driven DNA replication program of the human parasite Leishmania major. Cell Rep. 2016, 16, 1774–1786. [Google Scholar] [CrossRef]
- Martinez-Calvillo, S.; Romero-Meza, G.; Vizuet-de-Rueda, J.C.; Florencio-Martinez, L.E.; Manning-Cela, R.; Nepomuceno-Mejia, T. Epigenetic regulation of transcription in trypanosomatid protozoa. Curr. Genom. 2018, 19, 140–149. [Google Scholar] [CrossRef]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef]
- Koch, C.M.; Andrews, R.M.; Flicek, P.; Dillon, S.C.; Karaoz, U.; Clelland, G.K.; Wilcox, S.; Beare, D.M.; Fowler, J.C.; Couttet, P.; et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007, 17, 691–707. [Google Scholar] [CrossRef]
- Black, B.E.; Brock, M.A.; Bedard, S.; Woods, V.L., Jr.; Cleveland, D.W. An epigenetic mark generated by the incorporation of cenp-a into centromeric nucleosomes. Proc. Natl. Acad. Sci. USA 2007, 104, 5008–5013. [Google Scholar] [CrossRef]
- Quenet, D.; Dalal, Y. The CENP-A nucleosome: A dynamic structure and role at the centromere. Chromosome Res. 2012, 20, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Elwasila, M. Leishmania tarentolae wenyon, 1921 from the gecko Tarentola annularis in the Sudan. Parasitol. Res. 1988, 74, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Brun, R.; Schonenberger, M. Cultivation and in vitro cloning of procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979, 36, 289–292. [Google Scholar] [PubMed]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef]
- Assefa, S.; Keane, T.M.; Otto, T.D.; Newbold, C.; Berriman, M. Abacas: Algorithm-based automatic contiguation of assembled sequences. Bioinformatics 2009, 25, 1968–1969. [Google Scholar] [CrossRef]
- Otto, T.D.; Sanders, M.; Berriman, M.; Newbold, C. Iterative correction of reference nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 2010, 26, 1704–1707. [Google Scholar] [CrossRef]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Otto, T.D.; Dillon, G.P.; Degrave, W.S.; Berriman, M. RATT: Rapid annotation transfer tool. Nucleic Acids Res. 2011, 39, e57. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Stanke, M.; Schoffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Gassen, A.; Brechtefeld, D.; Schandry, N.; Arteaga-Salas, J.M.; Israel, L.; Imhof, A.; Janzen, C.J. DOT1a-dependent H3K76 methylation is required for replication regulation in Trypanosoma brucei. Nucleic Acids Res. 2012, 40, 10302–10311. [Google Scholar] [CrossRef]
- van Leeuwen, F.; Wijsman, E.R.; Kieft, R.; van der Marel, G.A.; van Boom, J.H.; Borst, P. Localization of the modified base J in telomeric VSG gene expression sites of Trypanosoma brucei. Genes Dev. 1997, 11, 3232–3241. [Google Scholar] [CrossRef][Green Version]
- Muller, L.S.M.; Cosentino, R.O.; Forstner, K.U.; Guizetti, J.; Wedel, C.; Kaplan, N.; Janzen, C.J.; Arampatzi, P.; Vogel, J.; Steinbiss, S.; et al. Genome organization and DNA accessibility control antigenic variation in trypanosomes. Nature 2018, 563, 121–125. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Location | Marker (s) | Chromatin State |
|---|---|---|
| Telomeres | H3.V, J | Unclear |
| TSSs | H2A.Z, H2B.V, H3K4me3, H3K16ac, H3K76me3 | Open |
| PTU body | H2A, H2B, H3 and H4 | Normal |
| TTSs | H3.V, J | Open |
| Centromere | H3K4me3, H3K16ac, H3K20me2, H3K36me3, (H3K50ac) * | Normal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDonald, J.R.; Jensen, B.C.; Sur, A.; Wong, I.L.K.; Beverley, S.M.; Myler, P.J. Localization of Epigenetic Markers in Leishmania Chromatin. Pathogens 2022, 11, 930. https://doi.org/10.3390/pathogens11080930
McDonald JR, Jensen BC, Sur A, Wong ILK, Beverley SM, Myler PJ. Localization of Epigenetic Markers in Leishmania Chromatin. Pathogens. 2022; 11(8):930. https://doi.org/10.3390/pathogens11080930
Chicago/Turabian StyleMcDonald, Jacquelyn R., Bryan C. Jensen, Aakash Sur, Iris L. K. Wong, Stephen M. Beverley, and Peter J. Myler. 2022. "Localization of Epigenetic Markers in Leishmania Chromatin" Pathogens 11, no. 8: 930. https://doi.org/10.3390/pathogens11080930
APA StyleMcDonald, J. R., Jensen, B. C., Sur, A., Wong, I. L. K., Beverley, S. M., & Myler, P. J. (2022). Localization of Epigenetic Markers in Leishmania Chromatin. Pathogens, 11(8), 930. https://doi.org/10.3390/pathogens11080930