
SUMOylation and Viral Infections of the Brain



Abstract
1. Introduction
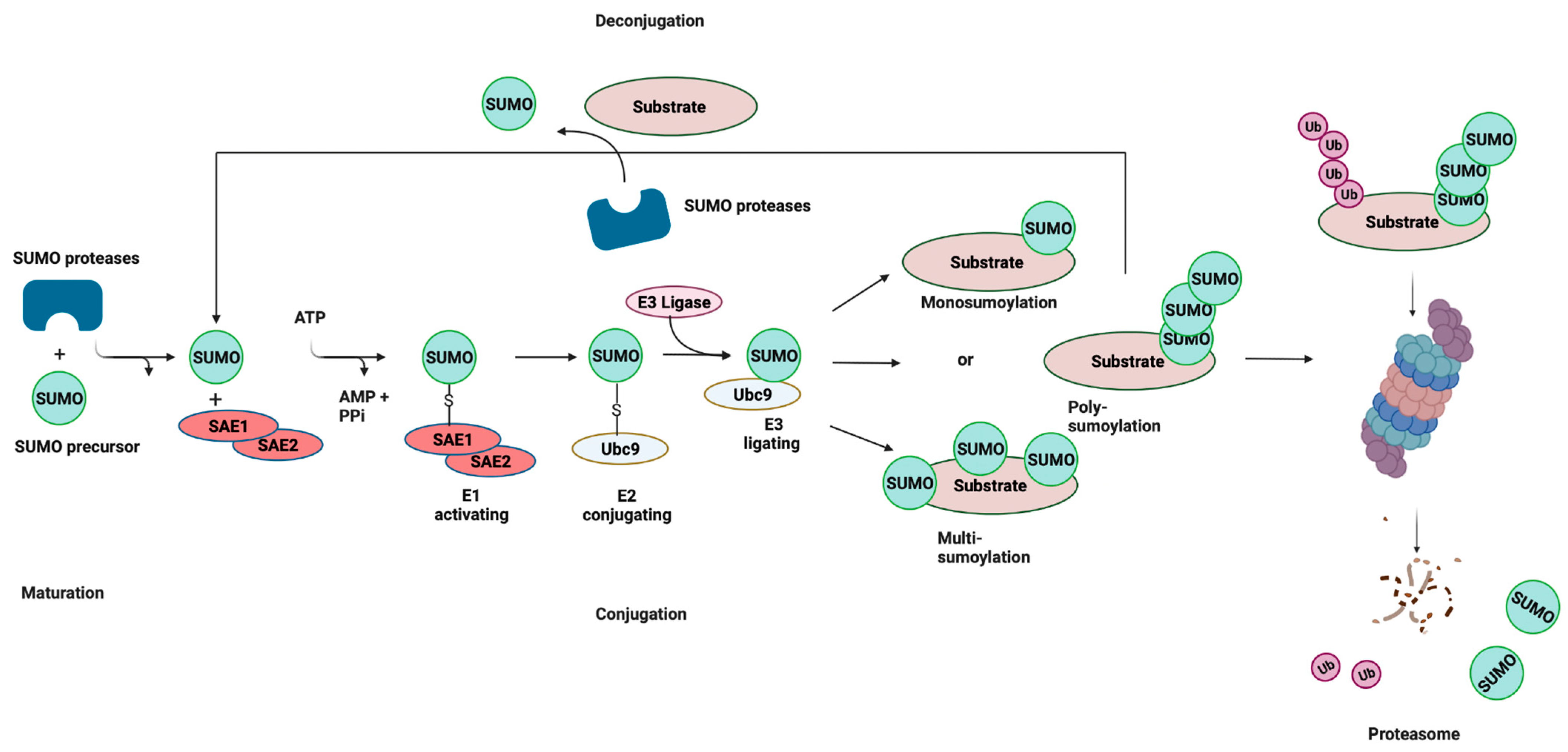
2. SUMOylation
3. SUMO Responses to Viral Infections
3.1. SUMO and HIV
3.2. SUMO and Coronaviruses
3.3. SUMO and Zika Virus
3.4. SUMO and HCMV
3.5. SUMO and Herpes Simplex Virus
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huber, A.K.; Irani, D.N. Is the Concept of Central Nervous System Immune Privilege Irrelevant in the Setting of Acute Infection? Front. Oncol. 2015, 5. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2015.00099/full (accessed on 13 September 2020). [CrossRef] [PubMed]
- Solomos, A.C.; Rall, G.F. Get It through Your Thick Head: Emerging Principles in Neuroimmunology and Neurovirology Redefine Central Nervous System Immune Privilege. ACS Chem. Neurosci. 2016, 7, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Bentz, G.L.; Shackelford, J.; Pagano, J.S. Epstein-Barr Virus Latent Membrane Protein 1 Regulates the Function of Interferon Regulatory Factor 7 by Inducing Its Sumoylation. J. Virol. 2012, 86, 12251. [Google Scholar] [CrossRef] [PubMed]
- Boggio, R.; Passafaro, A.; Chiocca, S. Targeting SUMO E1 to Ubiquitin Ligases: A viral strategy to counteract sumoylation*. J. Biol. Chem. 2007, 282, 15376–15382. [Google Scholar] [CrossRef]
- Boutell, C.; Cuchet-Lourenço, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence. PLoS Pathog. 2011, 7, e1002245. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3174244/ (accessed on 26 January 2021).
- Colombo, R.; Boggio, R.; Seiser, C.; Draetta, G.F.; Chiocca, S. The adenovirus protein Gam1 interferes with sumoylation of histone deacetylase 1. EMBO Rep. 2002, 3, 1062–1068. [Google Scholar] [CrossRef]
- Domingues, P.; Golebiowski, F.; Tatham, M.H.; Lopes, A.M.; Taggart, A.; Hay, R.T.; Hale, B.G. Global Reprogramming of Host SUMOylation during Influenza Virus Infection. Cell Rep. 2015, 13, 1467–1480. [Google Scholar] [CrossRef]
- Heaton, P.R.; Deyrieux, A.F.; Bian, X.L.; Wilson, V.G. HPV E6 proteins target Ubc9, the SUMO conjugating enzyme. Virus Res. 2011, 158, 199–208. [Google Scholar] [CrossRef]
- Johnson, E.S. Protein Modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef]
- Vertegaal, A.C.O.; Andersen, J.S.; Ogg, S.C.; Hay, R.T.; Mann, M.; Lamond, A.I. Distinct and Overlapping Sets of SUMO-1 and SUMO-2 Target Proteins Revealed by Quantitative Proteomics. Mol. Cell. Proteomics. 2006, 5, 2298–2310. [Google Scholar] [CrossRef]
- Talamillo, A.; Barroso-Gomila, O.; Giordano, I.; Ajuria, L.; Grillo, M.; Mayor, U.; Barrio, R. The role of SUMOylation during development. Biochem. Soc. Trans. 2020, 48, 463–478. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Li, M.; Zhang, Y.; Yang, P.; Eckenrode, S.; Hopkins, D.; Zheng, W.; Purohit, P.; Podolsky, P.H.; Muir, A.; et al. A functional variant of SUMO4, a new IκBα modifier, is associated with type 1 diabetes. Nat. Genet. 2004, 36, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell. Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Owerbach, D.; McKay, E.M.; Yeh, E.T.H.; Gabbay, K.H.; Bohren, K.M. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun. 2005, 337, 517–520. [Google Scholar] [CrossRef]
- Wei, W.; Yang, P.; Pang, J.; Zhang, S.; Wang, Y.; Wang, M.H.; Dong, Z.; She, J.-X.; Wang, C.-Y. A stress-dependent SUMO4 sumoylation of its substrate proteins. Biochem. Biophys. Res. Commun. 2008, 375, 454–459. [Google Scholar] [CrossRef]
- Kolli, N.; Mikolajczyk, J.; Drag, M.; Mukhopadhyay, D.; Moffatt, N.; Dasso, M.; Salvesen, G.; Wilkinson, K.D. Distribution and Paralog Specificity of Mammalian DeSUMOylating Enzymes. Biochem. J. 2010, 430, 335–344. [Google Scholar] [CrossRef]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef]
- Minty, A.; Dumont, X.; Kaghad, M.; Caput, D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J. Biol. Chem. 2000, 275, 36316–36323. [Google Scholar] [CrossRef]
- Song, J.; Durrin, L.K.; Wilkinson, T.A.; Krontiris, T.G.; Chen, Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 14373–14378. [Google Scholar] [CrossRef]
- Qu, Y.; Chen, Q.; Lai, X.; Zhu, C.; Chen, C.; Zhao, X.; Deng, R.; Xu, M.; Yuan, H.; Wang, Y.; et al. SUMOylation of Grb2 enhances the ERK activity by increasing its binding with Sos1. Mol. Cancer 2014, 13, 1–12. [Google Scholar] [CrossRef]
- Sarangi, P.; Zhao, X. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci. 2015, 40, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Mallin, D.R.; Simon, J.A.; Courey, A.J. Small ubiquitin-like modifier (SUMO) conjugation impedes transcriptional silencing by the polycomb group repressor Sex Comb on Midleg. J. Biol. Chem. 2011, 286, 11391–11400. [Google Scholar] [CrossRef] [PubMed]
- Princz, A.; Tavernarakis, N. SUMOylation in Neurodegenerative Diseases. Gerontology 2020, 66, 122–130. [Google Scholar] [CrossRef]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Imbert, F.; Langford, D. Viruses, SUMO, and immunity: The interplay between viruses and the host SUMOylation system. J. Neurovirol. 2021, 27, 531–541. [Google Scholar] [CrossRef]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; Straube, E.; et al. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef]
- Lescure, F.X.; Moulignier, A.; Savatovsky, J.; Amiel, C.; Carcelain, G.; Molina, J.M.; Gallien, S.; Pacanovski, J.; Pialoux, G.; Adle-Biassette, H.; et al. CD8 Encephalitis in HIV-Infected Patients Receiving cART: A Treatable Entity. Clin. Infect. Dis. 2013, 57, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 2016, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front Cell Infect Microbiol. 2019, 9. Available online: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00362/full (accessed on 16 September 2020). [CrossRef]
- Li, G.H.; Maric, D.; Major, E.O.; Nath, A. Productive HIV infection in astrocytes can be established via a nonclassical mechanism. AIDS 2020, 34, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLOS Pathog. 2020, 16, e1008381. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.A.; Zhao, M.L.; Lee, S.C. HIV-1 expression protects macrophages and microglia from apoptotic death. Neuropathol. Appl. Neurobiol. 2004, 30, 478–490. [Google Scholar] [CrossRef]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Zamborlini, A.; Coiffic, A.; Beauclair, G.; Delelis, O.; Paris, J.; Koh, Y.; Magne, F.; Giron, M.-L.; Tobaly-Tapiero, J.; Deprez, E.; et al. Impairment of Human Immunodeficiency Virus Type-1 Integrase SUMOylation Correlates with an Early Replication Defect*. J. Biol. Chem. 2011, 286, 21013–21022. [Google Scholar] [CrossRef] [PubMed]
- Gurer, C.; Berthoux, L.; Luban, J. Covalent modification of human immunodeficiency virus type 1 p6 by SUMO-1. J Virol. 2005, 79, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Vogel, W.K.; Liu, X.; Topark-Ngarm, A.; Arbogast, B.L.; Maier, C.S.; Filtz, T.M.; Leid, M. Coordinated regulation of transcription factor Bcl11b activity in thymocytes by the mitogen-activated protein kinase (MAPK) pathways and protein sumoylation. J. Biol. Chem. 2012, 287, 26971–26988. [Google Scholar] [CrossRef]
- Maison, C.; Bailly, D.; Quivy, J.P.; Almouzni, G. The methyltransferase Suv39h1 links the SUMO pathway to HP1α marking at pericentric heterochromatin. Nat Commun. 2016, 7, 12224. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell. 2007, 28, 823–837. [Google Scholar] [CrossRef]
- Li, M.; Xu, X.; Chang, C.W.; Liu, Y. TRIM28 functions as the SUMO E3 ligase for PCNA in prevention of transcription induced DNA breaks. Proc. Natl. Acad. Sci. USA 2020, 117, 23588–23596. [Google Scholar] [CrossRef]
- Liang, Q.; Deng, H.; Li, X.; Wu, X.; Tang, Q.; Chang, T.H.; Peng, H.; Rauscher, F.J.; Ozato, K.; Zhu, F. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 2011, 187, 4754–4763. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fiskus, W.; Yong, B.; Atadja, P.; Takahashi, Y.; Pandita, T.K.; Wang, H.-G.; Bhalla, K.N. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 6841–6846. [Google Scholar] [CrossRef]
- Gong, L.; Ji, W.K.; Hu, X.H.; Hu, W.F.; Tang, X.C.; Huang, Z.X.; Li, L.; Liu, M.; Xiang, S.-H.; Wu, E.; et al. Sumoylation differentially regulates Sp1 to control cell differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 5574–5579. [Google Scholar] [CrossRef]
- Marban, C.; Redel, L.; Suzanne, S.; Van Lint, C.; Lecestre, D.; Chasserot-Golaz, S.; Leid, M.; Aunis, D.; Schaeffer, E.; Rohr, O. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic. Acids. Res. 2005, 33, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Hamre, D.; Procknow, J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 1966, 121, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Chung, Y.S.; Jo, H.J.; Lee, N.J.; Kim, M.S.; Woo, S.H.; Park, S.; Kim, J.W.; Kim, H.M.; Han, M.-G. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res. Perspect. 2020, 11, 3. [Google Scholar] [CrossRef]
- Paraskevis, D.; Kostaki, E.G.; Magiorkinis, G.; Panayiotakopoulos, G.; Sourvinos, G.; Tsiodras, S. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol. 2020, 79, 104212. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 1–7. [Google Scholar] [CrossRef]
- Ding, Y.; He, L.I.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. J. Pathol. Soc. G. B. Irel. 2004, 203, 622–630. [Google Scholar] [CrossRef]
- Netland, J.; Meyerholz, D.K.; Moore, S.; Cassell, M.; Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008, 82, 7264–7275. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Sprado, A.; Skriabine, S.; Lu, P.; Weizman, O.-E.; Liu, F.; Dai, Y.; et al. Neuroinvasive potential of SARS-CoV-2 revealed in a human brain organoid model. J. Neurovirol. 2021, 27, 531–541. [Google Scholar]
- Kanberg, N.; Simrén, J.; Edén, A.; Andersson, L.M.; Nilsson, S.; Ashton, N.J.; Sundvall, P.-D.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Neurochemical signs of astrocytic and neuronal injury in acute COVID-19 normalizes during long-term follow-up. EBioMedicine 2021, 70, 103512. [Google Scholar] [CrossRef]
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D.; Al-Dalahmah, O.; Banu, M.A.; Boehme, A.K.; Boubour, A.L.; Bruce, S.S.; Chong, A.M.; Claassen, J.; et al. COVID-19 neuropathology at columbia university irving medical center/New York presbyterian hospital. Brain 2021, 144, 2696–2708. [Google Scholar] [CrossRef]
- Chang, R.Y.; Brian, D.A. cis Requirement for N-specific protein sequence in bovine coronavirus defective interfering RNA replication. J. Virol. 1996, 70, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Zhuo, Y.; Tan, X.; Zhou, Z.; Yuan, J.; Qiang, B.; Yan, J.; Peng, X.; Gao, G.F. SARS-CoV nucleocapsid protein binds to hUbc9, a ubiquitin conjugating enzyme of the sumoylation system. J. Med. Virol. 2006, 78, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Q.; Xiao, H.; Tam, J.P.; Liu, D.X. Sumoylation of the Nucleocapsid Protein of Severe Acute Respiratory Syndrome Coronavirus; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; Volume 579, pp. 2387–2396. Available online: https://febs.onlinelibrary.wiley.com/doi/abs/10.1016/j.febslet.2005.03.039 (accessed on 26 February 2021).
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on Zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef]
- Christian, K.M.; Song, H.; Ming, G.L. Pathophysiology and mechanisms of Zika virus infection in the nervous system. Annu. Rev. Neurosci. 2019, 42, 249–269. [Google Scholar] [CrossRef]
- Abdulaziz, A.T.A.; Zhou, D.; Li, J.M. Hydrocephalus in Guillain barre syndrome: A case report and review of the literature. Medicine 2020, 99, e18638. [Google Scholar] [CrossRef]
- Leonhard, S.E.; Bresani-Salvi, C.C.; Lyra Batista, J.D.; Cunha, S.; Jacobs, B.C.; Brito Ferreira, M.L.; Albuquerque, M.D.F.P.M.D. Guillain-Barré syndrome related to Zika virus infection: A systematic review and meta-analysis of the clinical and electrophysiological phenotype. PLoS Negl. Trop. Dis. 2020, 14, e0008264. [Google Scholar] [CrossRef]
- Gullo, G.; Scaglione, M.; Cucinella, G.; Riva, A.; Coldebella, D.; Cavaliere, A.F.; Signore, F.; Buzzaccarini, G.; Spagnol, G.; Laganà, A.S.; et al. Congenital Zika Syndrome: Genetic Avenues for Diagnosis and Therapy, Possible Management and Long-Term Outcomes. J. Clin. Med. 2022, 11, 1351. [Google Scholar] [CrossRef]
- Acosta-Ampudia, Y.; Monsalve, D.M.; Castillo-Medina, L.F.; Rodríguez, Y.; Pacheco, Y.; Halstead, S.; Willison, H.J.; Anaya, J.-M.; Ramírez-Santana, C. Autoimmune neurological conditions associated with Zika virus infection. Front. Mol. Neurosci. 2018, 11, 116. [Google Scholar] [CrossRef]
- Bido-Medina, R.; Wirsich, J.; Rodríguez, M.; Oviedo, J.; Miches, I.; Bido, P.; Tusen, L.; Stoeter, P.; Sadaghiani, S. Impact of Zika Virus on adult human brain structure and functional organization. Ann. Clin. Transl. Neurol. 2018, 5, 752–762. [Google Scholar] [CrossRef]
- Zhu, Z.; Chu, H.; Wen, L.; Yuan, S.; Chik, K.K.H.; Yuen, T.T.T.; Yip, C.C.-Y.; Wang, D.; Zhou, J.; Yin, F.; et al. Targeting SUMO Modification of the Non-Structural Protein 5 of Zika Virus as a Host-Targeting Antiviral Strategy. Int. J. Mol. Sci. 2019, 20, 392. [Google Scholar] [CrossRef] [PubMed]
- Elshahawi, H.; Syed Hassan, S.; Balasubramaniam, V. Importance of Zika virus NS5 protein for viral replication. Pathogens 2019, 8, 169. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.N.; Schutt, W.R.; Mladinich, M.; Sohn, S.Y.; Hearing, P.; Mackow, E.R. NS5 Sumoylation Directs Nuclear Responses That Permit Zika Virus To Persistently Infect Human Brain Microvascular Endothelial Cells. J. Virol. 2020, 94, e01086-20. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Maarifi, G.; McManus, F.P.; Lamoliatte, F.; Thibault, P.; Chelbi-Alix, M.K. Promyelocytic Leukemia Protein (PML) Requirement for Interferon-induced Global Cellular SUMOylation*. Mol. Cell. Proteomics. 2018, 17, 1196–1208. [Google Scholar] [CrossRef]
- Zhou, P.; Chen, X.; Li, M.; Tan, J.; Zhang, Y.; Yuan, W.; Zhou, J.; Wang, G. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem. Biophys. Res. Commun. 2019, 513, 1063–1069. [Google Scholar] [CrossRef]
- Schottstedt, V.; Blümel, J.; Burger, R.; Drosten, C.; Gröner, A.; Gürtler, L.; Heiden, M.; Hildebrandt, M.; Jansen, B.; Montag-Lessing, T.; et al. Human Cytomegalovirus (HCMV)-Revised. Transfus Med Hemotherapy Off Organ Dtsch Ges Transfusionsmedizin Immunhamatologie. Transfus. Med. Hemother. 2010, 37, 365–375. [Google Scholar]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- Lurain, N.S.; Chou, S. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 2010, 23, 689–712. [Google Scholar] [CrossRef]
- Brizić, I.; Lisnić, B.; Brune, W.; Hengel, H.; Jonjić, S. Cytomegalovirus infection: Mouse model. Curr. Protoc. Immunol. 2018, 122, e51. [Google Scholar] [CrossRef]
- Reuter, J.D.; Gomez, D.L.; Wilson, J.H.; Van Den Pol, A.N. Systemic immune deficiency necessary for cytomegalovirus invasion of the mature brain. J. Virol. 2004, 78, 1473–1487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krstanović, F.; Britt, W.J.; Jonjić, S.; Brizić, I. Cytomegalovirus Infection and Inflammation in Developing Brain. Viruses 2021, 13, 1078. [Google Scholar] [CrossRef] [PubMed]
- Lokensgard, J.R.; Cheeran, M.C.; Gekker, G.; Hu, S.; Chao, C.C.; Peterson, P.K. Human cytomegalovirus replication and modulation of apoptosis in astrocytes. J. Hum. Virol. 1999, 2, 91–101. [Google Scholar]
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Seiger, Å.; Söderberg-Nauclér, C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 2006, 80, 8929–8939. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Borysiewicz, L.K.; Morgan, B.P. Development of a model for cytomegalovirus infection of oligodendrocytes. J. Gen. Virol. 1997, 78, 3349–3356. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goodrum, F. Human cytomegalovirus latency: Approaching the Gordian knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef]
- Müller, S.; Dejean, A. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 1999, 73, 5137–5143. [Google Scholar] [CrossRef]
- Hwang, J.; Kalejta, R.F. Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J. Virol. 2009, 83, 6591–6598. [Google Scholar] [CrossRef]
- Sinigalia, E.; Alvisi, G.; Segré, C.V.; Mercorelli, B.; Muratore, G.; Winkler, M.; Hsiao, H.-H.; Urlaub, H.; Ripalti, A.; Chiocca, S.; et al. The Human Cytomegalovirus DNA Polymerase Processivity Factor UL44 Is Modified by SUMO in a DNA-Dependent Manner. PLoS ONE. 2012, 7, e49630. [Google Scholar] [CrossRef]
- Chen, J.; Li, G.; He, H.; Li, X.; Niu, W.; Cao, D.; Shen, A. Sumoylation of the carboxy-terminal of human cytomegalovirus DNA polymerase processivity factor UL44 attenuates viral DNA replication. Front. Microbiol. 2021, 12, 905. [Google Scholar] [CrossRef]
- Poole, E.L.; Kew, V.G.; Lau, J.C.; Murray, M.J.; Stamminger, T.; Sinclair, J.H.; Reeves, M.B. A virally encoded DeSUMOylase activity is required for cytomegalovirus reactivation from latency. Cell. Rep. 2018, 24, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A.V.; Hou, J.; Major, E.O.; Straus, S.E. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005, 79, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J. Herpes simplex virus infection. In Seminars in Pediatric Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2002; pp. 6–11. [Google Scholar]
- Liesegang, T.J. Herpes simplex virus epidemiology and ocular importance. Cornea 2001, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef]
- Whitbeck, J.C.; Peng, C.; Lou, H.; Xu, R.; Willis, S.H.; de Leon, M.P.; Peng, T.; Nicola, A.V.; I Montgomery, R.; Warner, M.S.; et al. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J Virol. 1997, 71, 6083–6093. [Google Scholar] [CrossRef]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRα is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin-and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell. Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef]
- Clement, C.; Tiwari, V.; Scanlan, P.M.; Valyi-Nagy, T.; Yue, B.Y.; Shukla, D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell. Biol. 2006, 174, 1009–1021. [Google Scholar] [CrossRef]
- Marques, C.P.; Hu, S.; Sheng, W.; Lokensgard, J.R. Microglial cells initiate vigorous yet non-protective immune responses during HSV-1 brain infection. Virus Res. 2006, 121, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schachtele, S.J.; Hu, S.; Little, M.R.; Lokensgard, J.R. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflammation 2010, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Müller, S.; Ronchetti, S.; Freemont, P.S.; Dejean, A.; Pandolfi, P.P. Role of SUMO-1–modified PML in nuclear body formation. Blood 2000, 95, 2748–2752. [Google Scholar] [CrossRef]
- Boutell, C.; Everett, R.D. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen Virol. 2013, 94, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Orr, A.; Everett, R.D. PML Residue Lysine 160 Is Required for the Degradation of PML Induced by Herpes Simplex Virus Type 1 Regulatory Protein ICP0. J. Virol. 2003, 77, 8686–8694. [Google Scholar] [CrossRef]
- Sloan, E.; Tatham, M.H.; Groslambert, M.; Glass, M.; Orr, A.; Hay, R.T.; Everett, R.D. Analysis of the SUMO2 Proteome during HSV-1 Infection. PLoS Pathog. 2015, 11, e1005059. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Protein | SUMO Protein | SUMOylation Interactions | References |
|---|---|---|---|---|
| HIV | Integrase (IN) | SUMO1 and SUMO2/3 | Inhibits viral genome integration | [37] |
| P6 | SUMO1 | Reduces infectivity of released virions | [38] | |
| SARS-CoV-1 | NP | SUMO1 | Promotes homo-oligomerization | [64] |
| HCMV | IE1 | SUMO1 | Abrogates interaction of SUMO1 with PML | [89] |
| pp71 | SUMO1 | Promotes SUMOylation of Daxx | [90] | |
| UL44 | SUMO1 and SUMO2/3 | Enhances virus production and DNA replication | [91] | |
| SUMO1 and SUMO2/3 | Attenuates virus production | [92] | ||
| LUNA | N/A | DeSUMOylates PML | [93] | |
| Zika | NS5 | SUMO1 | SUMO1 conjugation stabilizes NS5 | [76] |
| HSV-1 | ICP0 | N/A | DeSUMOylates PML and induces it degradation | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imbert, F.; Leavitt, G.; Langford, D. SUMOylation and Viral Infections of the Brain. Pathogens 2022, 11, 818. https://doi.org/10.3390/pathogens11070818
Imbert F, Leavitt G, Langford D. SUMOylation and Viral Infections of the Brain. Pathogens. 2022; 11(7):818. https://doi.org/10.3390/pathogens11070818
Chicago/Turabian StyleImbert, Fergan, Gabrielle Leavitt, and Dianne Langford. 2022. "SUMOylation and Viral Infections of the Brain" Pathogens 11, no. 7: 818. https://doi.org/10.3390/pathogens11070818
APA StyleImbert, F., Leavitt, G., & Langford, D. (2022). SUMOylation and Viral Infections of the Brain. Pathogens, 11(7), 818. https://doi.org/10.3390/pathogens11070818