
Genome Analysis of ESBL-Producing Escherichia coli Isolated from Pigs

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Baseline Characteristics and Phenotypic Analyses
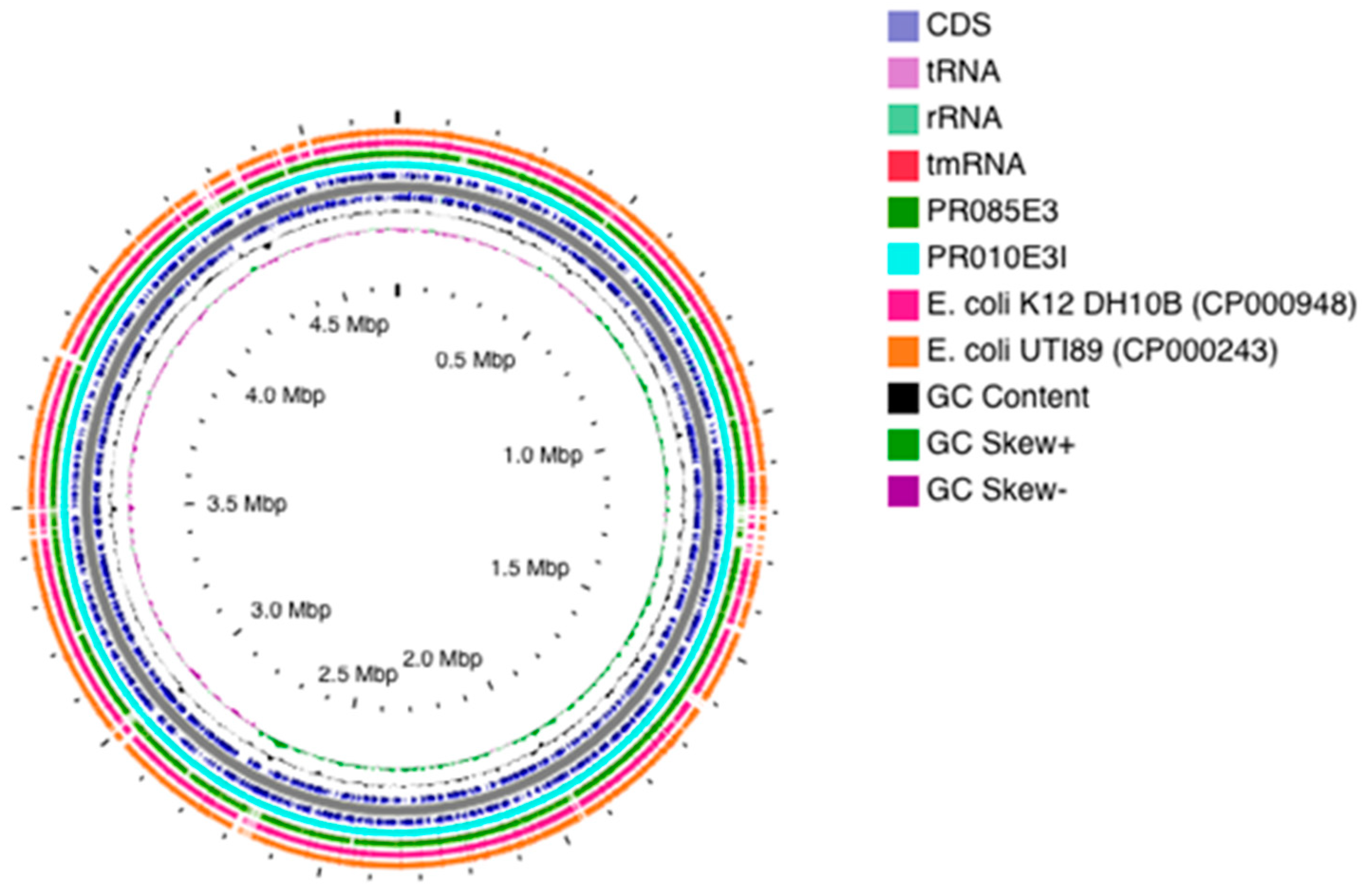
2.2. Genomic Features
2.3. Antimicrobial Resistance Phenotypes and Genotypes
2.4. Whole-Genome Virulome Profiling and Pathogenicity
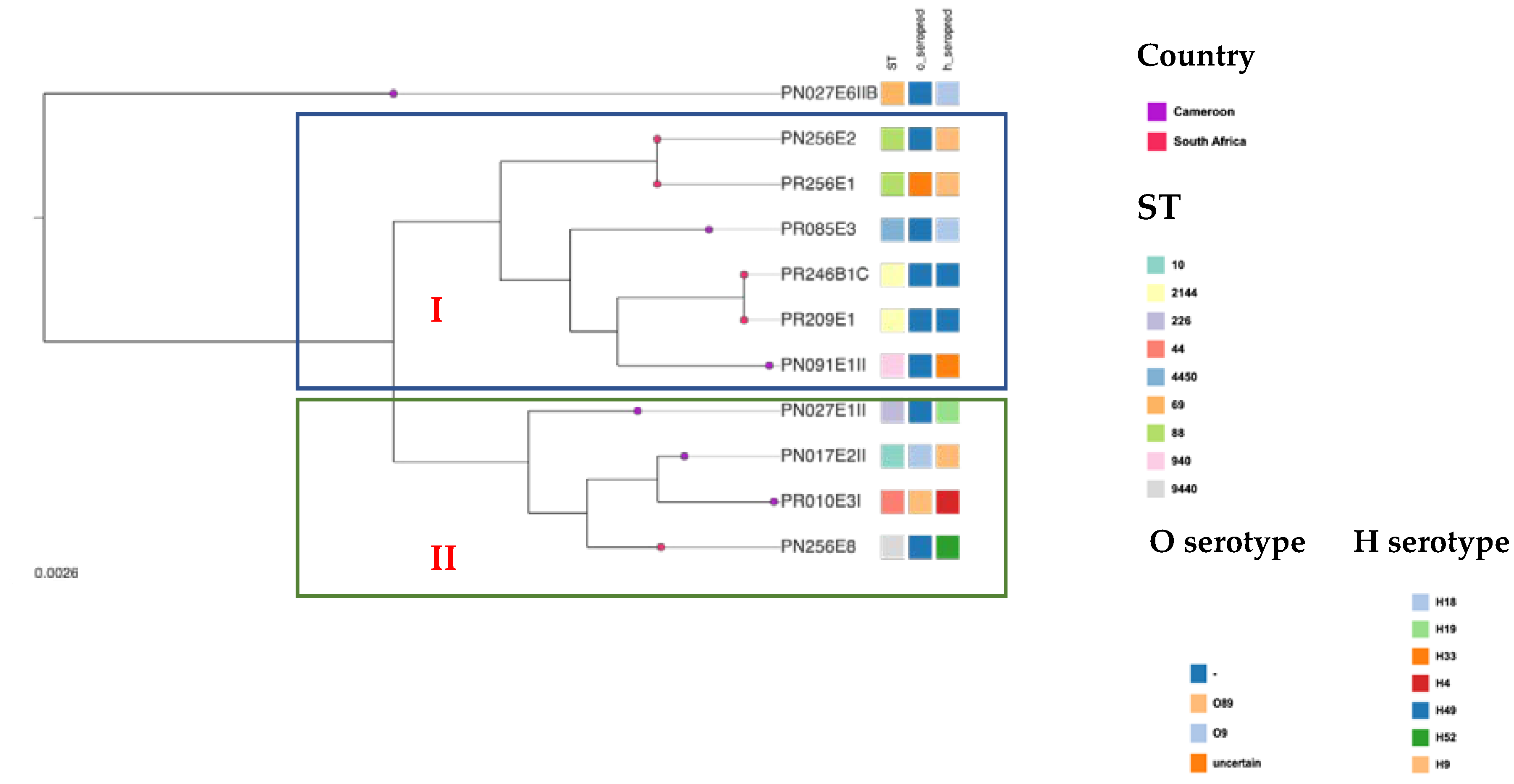
2.5. Phylogenetic Groups and Multilocus Sequence Typing, Serotyping and Phylotyping
2.6. Mobile Genetic Elements
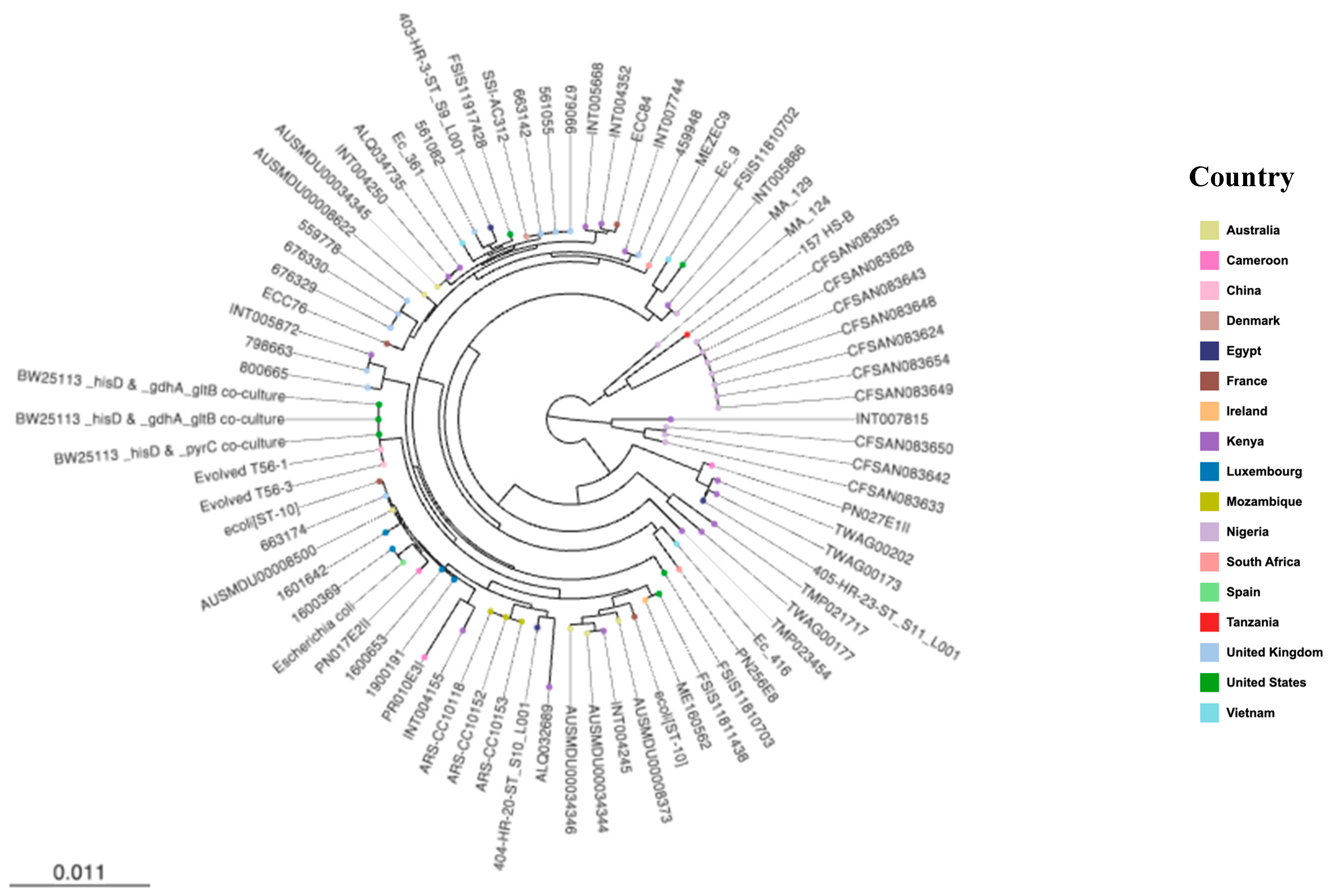
2.7. Phylogenetic Analysis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Study Design and Bacterial Isolates
5.2. Identification, ESBL Screening and Antimicrobial Susceptibility Testing
5.3. Whole Genome Sequencing and Data Analysis
5.3.1. Purification, Sequencing and Pre-Processing of Genomic Data
5.3.2. WGS-Based Molecular Typing
5.3.3. In Silico Resistome and Virulome Profiling
5.3.4. Detection of Mobile Genetic Elements
5.3.5. Genome Visualization and Gene Annotation
5.3.6. Comparative Phylogenomic Analyses
5.4. Nucleotide Accession Number
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; The Review on Antimicrobial Resistance: London, UK, 2016. [Google Scholar]
- Founou, L.L.; Founou, R.C.; Essack, S.Y. Antibiotic Resistance in the Food Chain: A Developing Country-Perspective. Front. Microbiol. 2016, 7, 1881. [Google Scholar] [CrossRef] [PubMed]
- Sarowska, J.; Futoma-Koloch, B.; Jama-Kmiecik, A.; Frej-Madrzak, M.; Ksiazczyk, M.; Bugla-Ploskonska, G.; Choroszy-Krol, I. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: Recent reports. Gut Pathog. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selander, R.K.; Caugant, D.A.; Whittam, T.S. Genetic Structure and Variation in Natural Populations of Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology; Ingraham, J.L., Magasanik, B., Schaechter, M., Low, K.B., Neidhardt, F.C., Umbarger, H.E., Eds.; American Society for Microbiology: Washington, DC, USA, 1987; pp. 1625–1648. [Google Scholar]
- Herzer, P.J.; Inouye, S.; Inouye, M.; Whittam, T.S. Phylogenetic distribution of branched RNA-linked multicopy single-stranded DNA among natural isolates of Escherichia coli. J. Bacteriol. 1990, 172, 6175–6181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perovic, O.; Singh-Moodley, A.; Dusé, A.; Bamford, C.; Elliott, G.; Han, K.S.S.; Kularatne, R.; Lowman, W.; Whitelaw, A.; Nana, T.; et al. National sentinel site surveillance for antimicrobial resistance in Klebsiella pneumoniae isolates in South Africa, 2010–2012. S. Afr. Med. J. 2014, 104, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Founou, L.L.; Founou, R.C.; Essack, S.Y. Antimicrobial resistance in the farm-to-plate continuum: More than a food safety issue. Futur. Sci. OA 2021, 7, FSO692. [Google Scholar] [CrossRef]
- Chishimba, K.; Hang’ombe, B.M.; Muzandu, K.; Mshana, S.E.; Matee, M.I.; Nakajima, C.; Suzuki, Y. Detection of Extended-Spectrum Beta-Lactamase-Producing Escherichia coli in Market-Ready Chickens in Zambia. Int. J. Microbiol. 2016, 2016, 5275724. [Google Scholar] [CrossRef] [Green Version]
- Aworh, M.K.; Kwaga, J.; Okolocha, E.; Harden, L.; Hull, D.; Hendriksen, R.S.; Thakur, S. Extended-spectrum ß-lactamase-producing Escherichia coli among humans, chickens and poultry environments in Abuja, Nigeria. One Health Outlook 2020, 2, 8. [Google Scholar] [CrossRef]
- Runcharoen, C.; Raven, K.E.; Reuter, S.; Kallonen, T.; Paksanont, S.; Thammachote, J.; Anun, S.; Blane, B.; Parkhill, J.; Peacock, S.J.; et al. Whole genome sequencing of ESBL-producing Escherichia coli isolated from patients, farm waste and canals in Thailand. Genome Med. 2017, 9, 81. [Google Scholar] [CrossRef]
- Hoek, A.H.A.M.V.; Dierikx, C.; Bosch, T.; Schouls, L.; Van Duijkeren, E.; Visser, M. Transmission of ESBL-producing Escherichia coli between broilers and humans on broiler farms. J. Antimicrob. Chemother. 2020, 75, 543–549. [Google Scholar] [CrossRef]
- Founou, L.L.; Founou, R.C.; Ntshobeni, N.; Govinden, U.; Bester, L.A.; Chenia, H.Y.; Djoko, C.F.; Essack, S.Y. Emergence and Spread of Extended Spectrum β-Lactamase Producing Enterobacteriaceae (ESBL-PE) in Pigs and Exposed Workers: A Multicentre Comparative Study between Cameroon and South Africa. Pathogens 2019, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Mathers, A.J.; Peirano, G.; Pitout, J.D.D. The Role of Epidemic Resistance Plasmids and International High-Risk Clones in the Spread of Multidrug-Resistant Enterobacteriaceae. Clin. Microbiol. Rev. 2015, 28, 565–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafaï, C.; Frank, T.; Manirakiza, A.; Gaudeuille, A.; Mbecko, J.-R.; Nghario, L.; Serdouma, E.; Tekpa, B.; Garin, B.; Breurec, S. Dissemination of IncF-type plasmids in multiresistant CTX-M-15-producing Enterobacteriaceae isolates from surgical-site infections in Bangui, Central African Republic. BMC Microbiol. 2015, 15, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbelle, N.M.; Feldman, C.; Sekyere, J.O.; Maningi, N.E.; Modipane, L.; Essack, S.Y. The Resistome, Mobilome, Virulome and Phylogenomics of Multidrug-Resistant Escherichia coli Clinical Isolates from Pretoria, South Africa. Sci. Rep. 2019, 9, 16457. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.M.; McDermott, P.F. Using Genomics to Track Global Antimicrobial Resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammad, A.M.; Hoffmann, M.; Gonzalez-Escalona, N.; Abbas, N.H.; Yao, K.; Koenig, S.; Allué-Guardia, A.; Eppinger, M. Genomic features of colistin resistant Escherichia coli ST69 strain harboring mcr-1 on IncHI2 plasmid from raw milk cheese in Egypt. Infect. Genet. Evol. 2019, 73, 126–131. [Google Scholar] [CrossRef]
- Li, B.; Sun, J.-Y.; Han, L.-Z.; Huang, X.-H.; Fu, Q.; Ni, Y.-X. Phylogenetic Groups and Pathogenicity Island Markers in Fecal Escherichia coli Isolates from Asymptomatic Humans in China. Appl. Environ. Microbiol. 2010, 76, 6698–6700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoppe, N.D.C.; Silva, J.S.; Carlos, C.; Sato, M.I.Z.; Saraiva, A.M.; Ottoboni, L.M.M.; Torres, T.T. Worldwide Phylogenetic Group Patterns of Escherichia coli from Commensal Human and Wastewater Treatment Plant Isolates. Front. Microbiol. 2017, 8, 2512. [Google Scholar] [CrossRef] [Green Version]
- Olowe, O.A.; Adefioye, O.J.; Ajayeoba, T.A.; Schiebel, J.; Weinreich, J.; Ali, A.; Burdukiewicz, M.; Rödiger, S.; Schierack, P. Phylogenetic grouping and biofilm formation of multidrug resistant Escherichia coli isolates from humans, animals and food products in South-West Nigeria. Sci. Afr. 2019, 6, e00158. [Google Scholar] [CrossRef]
- Tadesse, D.A.; Zhao, S.; Tong, E.; Ayers, S.; Singh, A.; Bartholomew, M.J.; McDermott, P.F. Antimicrobial Drug Resistance in Escherichia coli from Humans and Food Animals, United States, 1950–2002. Emerg. Infect. Dis. 2012, 18, 741–749. [Google Scholar] [CrossRef]
- Patil, S.; Chen, X.; Lian, M.; Wen, F. Phenotypic and genotypic characterization of multi-drug-resistant Escherichia coli isolates harboring blaCTX-M group extended-spectrum β-lactamases recovered from pediatric patients in Shenzhen, southern China. Infect. Drug Resist. 2019, 12, 1325–1332. [Google Scholar] [CrossRef] [Green Version]
- EUCAST. Breakpoint Tables for Interpretation of MICs and Zone Diameters. 2016. Available online: https://www.eucast.org/ (accessed on 28 March 2017).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alikhan, N.-F.; Zhou, Z.; Sergeant, M.J.; Achtman, M. A genomic overview of the population structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, O.; Christenson, J.K.; Denamur, E.; Gordon, D.M. The Clermont Escherichia coli phylo-typing method revisited: Improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013, 5, 58–65. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2018, 47, D687–D692. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.-A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis comparison tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, M.G.; Li, M.M.; Siranosian, B.A.; Montgomery, S.B.; Bhatt, A.S. A Bioinformatic Analysis of Integrative Mobile Genetic Elements Highlights Their Role in Bacterial Adaptation. Cell Host Microbe 2020, 27, 140–153.e149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids. Antimicrob using PlasmidFinder and plasmid multilocus sequence typing. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.R.; Arantes, A.S.; Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genom. 2012, 13, 202. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Alikhan, N.-F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Accession Number | Country | Sample Type | Abattoir | MLST * | Clonal Complex | FimH | Phylogroup | Serotype |
|---|---|---|---|---|---|---|---|---|---|
| PN017E2II | VMKK00000000 | Cameroon | Nasal swab | SH001 | 10 | ST10 Cplx | FimH215 | A | O9:H:9 |
| PR010E3I | VKOQ00000000 | Cameroon | Rectal swab | SH001 | 44 | ST10 Cplx | FimH54 | A | O89:H4 |
| PN027E6IIB | VKOV00000000 | Cameroon | Nasal swab | SH001 | 69 | ST69 Cplx | FimH27 | D | O-:H18 |
| PR256E1 | VKOS00000000 | South Africa | Rectal swab | SH005 | 88 | ST23 Cplx | FimH1250 | C | O: Uncertain H9 |
| PN256E2 | VKOT00000000 | South Africa | Nasal swab | SH005 | 88 | ST23 Cplx | FimH1250 | C | O-:H9 |
| PN027E1II | VKOW00000000 | Cameroon | Nasal swab | SH001 | 226 | ST226 Cplx | FimH43 | A | O-:H19 |
| PN091E1II | VKOU00000000 | Cameroon | Nasal swab | SH002 | 940 | ST448 Cplx | Unknown | B1 | O-:H33 |
| PN256E8 | QJRZ00000000 | South Africa | Nasal swab | SH005 | 9440 | ST10 Cplx | FimH23 | A | O-:H52 |
| PR209E1 | VKOO00000000 | South Africa | Rectal swab | SH004 | 2144 | - | FimH87 | B1 | O-:H49 |
| PR246B1C | WHRW00000000 | South Africa | Rectal swab | SH004 | 2144 | - | FimH87 | B1 | O-:H49 |
| PR085E3 | VKOP000000000 | Cameroon | Rectal swab | SH002 | 4450 | - | FimH566 | A | O-:H18 |
| Isolate | Country | Sample Type | Abattoir | MLST # | β-Lactamase Resistance Genes | Fluoroquinolone Resistance Genes | Other Resistance Genes | Plasmids | pMLST * | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTX-M | TEM | OXA | QRDR | PMQR | ||||||||
| PN017E2II | Cameroon | Nasal swab | SH001 | 10 | CTX-M-15 | TEM-1B | - | - | qnrS1 | aph(6)-Id, aph(3″)-Ib, tet(A), mph(A), sul2, dfrA14, | IncY, Col(MG828), Col440I, rep21 | - |
| PR010E3I | Cameroon | Rectal swab | SH001 | 44 | CTX-M-15 | - | OXA-1 | gyrA (p.S83L) gyrA (p.D87N) | aac(6′)-Ib-cr | aac(3)-IIa, aph(3″)-Ib, aadA5, aph(6)-Id, tet(B), tet(A), sul1, sul2, dfrA17, floR, catB3 | IncFIA, Col440I, IncFII, IncFIB, Col(MG828), rep21 | IncF [F36:A20:B1] |
| PN027E6IIB | Cameroon | Nasal swab | SH001 | 69 | CTX-M-15 | TEM-1B | - | - | qnrS1 | strA, strB, sul2, tet(A), dfrA14 | IncY, Col(MG828) | - |
| PR256E1 | South Africa | Rectal swab | SH005 | 88 | CTX-M-1 | - | - | - | - | tet(A), sul2, | IncI1 &, IncI2, Col(MG828), ColPVC, IncFIB, | IncF [K-:A-:B1]; IncI1[ST3] |
| PN256E2 | South Africa | Nasal swab | SH005 | 88 | CTX-M-1 | - | - | - | - | tet(A), sul2, | IncI1 **, IncFIB, Col(MG828), Col440I, rep10 | IncF [K-:A-:B1]; IncI1[ST3] |
| PN027E1II | Cameroon | Nasal swab | SH001 | 226 | CTX-M-15 | TEM-1B | - | - | qnrS1 | aph(3″)-Ib, aph(6)-Id, tet(A), mdf(A), sul2, dfrA14, | IncY, Col440I, colRNAI, Col(MG828) | - |
| PN091E1II | Cameroon | Nasal swab | SH002 | 940 | CTX-M-15 | TEM-1B | - | gyrA (p.S83A), | - | aph(3″)-Ib, aph(6)-Id, aadA1, 16S_rrsC (g.926_926del), tet(B), mph(A), sul2, dfrA1, | IncX, Col440I | - |
| PN256E8 | South Africa | Nasal swab | SH005 | 944 | CTX-M-55 | TEM-1B TEM-141 TEM-206 | - | - | oqxA, oqxB aac(6′)-Ib-cr | aac(6′)-Ib3, aadA5, tet(A), sul2, dfrA17, floR, mcr-1.1, fosA3 | IncN, IncHI2A, IncHI2 | IncN [ST1]; IncHI2 [ST3-like] |
| PR209E1 | South Africa | Rectal swab | SH004 | 2144 | CTX-M-14 | - | - | - | oqxB, oqxA | aph(3″)-Ib, aph(6)-Id, aadA2b, aadA1, sul3, cmlA1, fosA3 | IncFIC(FII), IncFIB, IncHI2A, IncHI2 rep21 | IncF [K89:A-:B57] IncHI2[ST3] |
| PR246B1C | South Africa | Rectal swab | SH004 | 2144 | CTX-M-14 | - | - | - | oqxA, oqxB | aph(3″)-Ib, aadA2b, aph(6)-Id, aadA1, aph(3″)-Ib, sul3, fosA3, cmlA1 | IncFIC(FII), Col440II, IncHI2A, IncHI2, IncFIB | IncF [K89:A-:B57] IncHI2 [ST3] |
| PR085E3 | Cameroon | Rectal swab | SH002 | 4450 | CTX-M-15 | - | - | - | qnrS1 | AadA5, sul2, dfrA17 | IncY | - |
| Pathogenicity Feature | Nasal Isolates | Rectal Isolates | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PN017E2II | PN027E6IIB | PN027E1II | PN091E1II | PN256E2 | PN256E8 | PR010E3I | PR209E1 | PR246B1C | PR256E1 | PR085E3 | |
| Pathogenicity Score (No. of Pathogenic Families) | 0.934 (615) | 0.937 (889) | 0.94 (526) | 0.941 (665) | 0.927 (735) | 0.932 (625) | 0.94 (677) | 0.939 (710) | 0.937 (682) | 0.929 (729) | 0.939 (666) |
| Human Pathogenicity | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Virulence Factors | |||||||||||
| Adherence | ecp, elf, eae, hcp, fim | lpfA, | elf, hcp, fim | cfa, ecp, elf, eae, hcp, lpfA | hra, lpfA, tsh, cfa, ecp, elf, eae, foc, hcp, pap, fim, pil | - | hra, papA_F19, ecp, elf, eae, hcp, papI, fim, | lpfA, cfa, ecp, elf, eae, hcp, papI, fim | lpfA | pap, foc, lpfA, tsh, | hra, lpfA, pap, tsh, cfa, ecp, elf, eae, foc, hcp, fim, lfhA, prl/gapA, cgs, pilW, sta, stf, stgB |
| Autotransporter | EhaB | - | aatA, ehaB, upaG/ehaG | ehaB, upaG/ehaG | agn43, ehaB, upaG/ehaG, | - | cah, ehaB | eha, upaG/ehaG, | - | - | cah, ehaAB, upaG/ehaG, |
| Iron Uptake | - | fyuA, irp, sitA, | - | fyuA, irp, ybt | iuc, iut, sitABC, iro, iroN, fyuA, irp, ybt | - | iut, sitABCD | - | - | irp, iuc, iutA, iroN, fyuA | iroN, ccmF, ent, fep, hem |
| Secretion system | aec | - | - | aec | aec | - | aec | aec | - | etsC | aec, flg, flh, fli, ipaH, gsp, clpB |
| Antiphagocytosis | wzc, wzi | - | - | wzc, wzi, wbaZ | - | - | wzi | - | - | - | rmkB, wbjD/wecB, wecC, galF, ugd, wcal, wzc |
| Toxins | hlyE | - | - | - | hlyF, astA, vat | astA | hlyE | hlyE | - | hlyF, | hlyAE |
| Protectins and invasins | ibeBC | KpsE, kpsMIII_K96; iss, ompT | ibeBC | iss ibeBC | KpsE, kpsMIII_K96, iss, ompT, traT, ibeBC | ompT, | traT, ibeBC, tia | iss, ompT, ibeBC, | traT, ompT, iss, | iss, ompT, traT, | ibeBC, che, motA, |
| Miscellaneous | espL espX, galE, rmlD, gad, terC | air, terC, gad, chuA, eilA | espL, espX, rmlD | espL, espX, terC, gad, | mch, mcmA, terC, gad, eilA, air | terC, gad, | esp, gad, terC, rmlD, galE, cea | esp, terC, gad, adeG, air | gad, terC | cea, cib, mch, mcmA, terC | esp, gal, mrsA/glmM, pgi, acpXL, rml, rpoS, phoQ, glnA1, narH, sugC, acrB, farB, icl, mgtB, motB, bioB, katG, gmhA/lpcA, htrB, kdsA, kdtA, lpxABK, msbA, opsX/rfaC, rfa, wecA, air |
| Isolate (ST) | Plasmids | Insertion Sequence | Transposons | Phages | CRISPR Array (Cas System) | TR |
|---|---|---|---|---|---|---|
| PN017E2II (10) | IncY, Col(MG828), Col440I, rep21 | - | - | - | 6 (Cas1) | 54 |
| PR010E3I (44) | IncFIA, Col440I, IncFII, IncFIB, Col(MG828), rep21 | - | - | - | 8 (Cas1, Cas3) | 48 |
| PN027E6IIB (69) | IncY, Col(MG828) | ISKpn19, ISEc1, ISEc31, IS4, ISSfl10, IS911, cn_5813_IS911, MITEEc1, ISEc38, IS629, ISEc46, IS5075 | - | PHAGE_Entero_mEp460_NC_019716 | 5 (Cas1) | 54 |
| PR256E1 (88) | IncI1 &, IncI2, Col(MG828), ColPVC, IncFIB, | IS26, ISVsa3, ISSbo1, cn_3792_ISSbo1, ISEc9, ISEc40, ISEc38, ISEc13 | - | PHAGE_Entero_fiAA91_ss_NC_022750 PHAGE_Shigel_SfII_NC_021857(34) PHAGE_Entero_HK544_NC_019767 | 6 (Cas2) | 51 |
| PN256E2 (88) | IncI1 *, IncFIB, Col(MG828), Col440I, rep10 | IS26, ISVsa3, ISEc9 | - | - | 10 (Cas3) | 101 |
| PN027E1II (226) | IncY, Col440I, colRNAI, Col(MG828) | ISKpn19, ISEsa1, IS5075, MITEEc1, IS100, ISEc30, IS5, ISEc26, ISKpn8, IS421, IS609, ISEc38, IS30, IS903 | - | - | 11 (Cas3, Cas1) | 55 |
| PN091E1II (940) | IncX, Col440I | IS6100, MITEEc1, IS421, ISEc30, ISSfl10, IS30, ISEc38, ISEc1, IS100, ISKpn8 | Tn7 # | PHAGE_Entero_BP_4795_NC_004813 | 5 (Cas2) | 40 |
| PN256E8 (944) | IncN, IncHI2A, IncHI2 | ISVsa3, IS640, IS100, ISEam1, IS30, MITEEc1, ISEc1, ISKpn26, IS421, ISVsa5, IS609 | - | PHAGE_Shigel_SfII_NC_021857(34) | 8 (Cas2) | 87 |
| PR209E1 (2144) | IncFIC(FII), IncFIB, IncHI2A, IncHI2 rep21 | IS102, IS629, MITEEc1, ISKpn8, ISVsa5, IS421, IS3, IS26 | Tn6082 | PHAGE_Shigel_Sf6_NC_005344 PHAGE_Shigel_Sf6_NC_005344 PHAGE_Shigel_SfII_NC_021857(34) | 6 (Cas2) | 44 |
| PR246B1C (2144) | IncFIC(FII), Col440II, IncHI2A, IncHI2, IncFIB | IS102, IS3, IS629, IS26, ISEc1, ISKpn8, ISVsa5, IS421, MITEEc1 | Tn6082 | PHAGE_Shigel_Sf6_NC_005344 PHAGE_Shigel_SfII_NC_021857 PHAGE_Entero_fiAA91_ss_NC_022750 | 8 | 41 |
| PR085E3 (4450) | IncY | ISVsa3, ISEc9, IS421, ISKpn26, IS3, ISEc1, ISEc38, MITEEc1, IS26, IS102 | - | PHAGE_Entero_mEp460_NC_019716 PHAGE_Pseudo_phiPSA1_NC_024365 PHAGE_Entero_fiAA91_ss_NC_022750 | 4 (Cas3) | 39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Founou, L.L.; Founou, R.C.; Allam, M.; Ismail, A.; Essack, S.Y. Genome Analysis of ESBL-Producing Escherichia coli Isolated from Pigs. Pathogens 2022, 11, 776. https://doi.org/10.3390/pathogens11070776
Founou LL, Founou RC, Allam M, Ismail A, Essack SY. Genome Analysis of ESBL-Producing Escherichia coli Isolated from Pigs. Pathogens. 2022; 11(7):776. https://doi.org/10.3390/pathogens11070776
Chicago/Turabian StyleFounou, Luria Leslie, Raspail Carrel Founou, Mushal Allam, Arshad Ismail, and Sabiha Yusuf Essack. 2022. "Genome Analysis of ESBL-Producing Escherichia coli Isolated from Pigs" Pathogens 11, no. 7: 776. https://doi.org/10.3390/pathogens11070776
APA StyleFounou, L. L., Founou, R. C., Allam, M., Ismail, A., & Essack, S. Y. (2022). Genome Analysis of ESBL-Producing Escherichia coli Isolated from Pigs. Pathogens, 11(7), 776. https://doi.org/10.3390/pathogens11070776