
Surgical Strikes on Host Defenses: Role of the Viral Protease Activity in Innate Immune Antagonism


{kind=link}
{kind=link}
Abstract
:1. Introduction
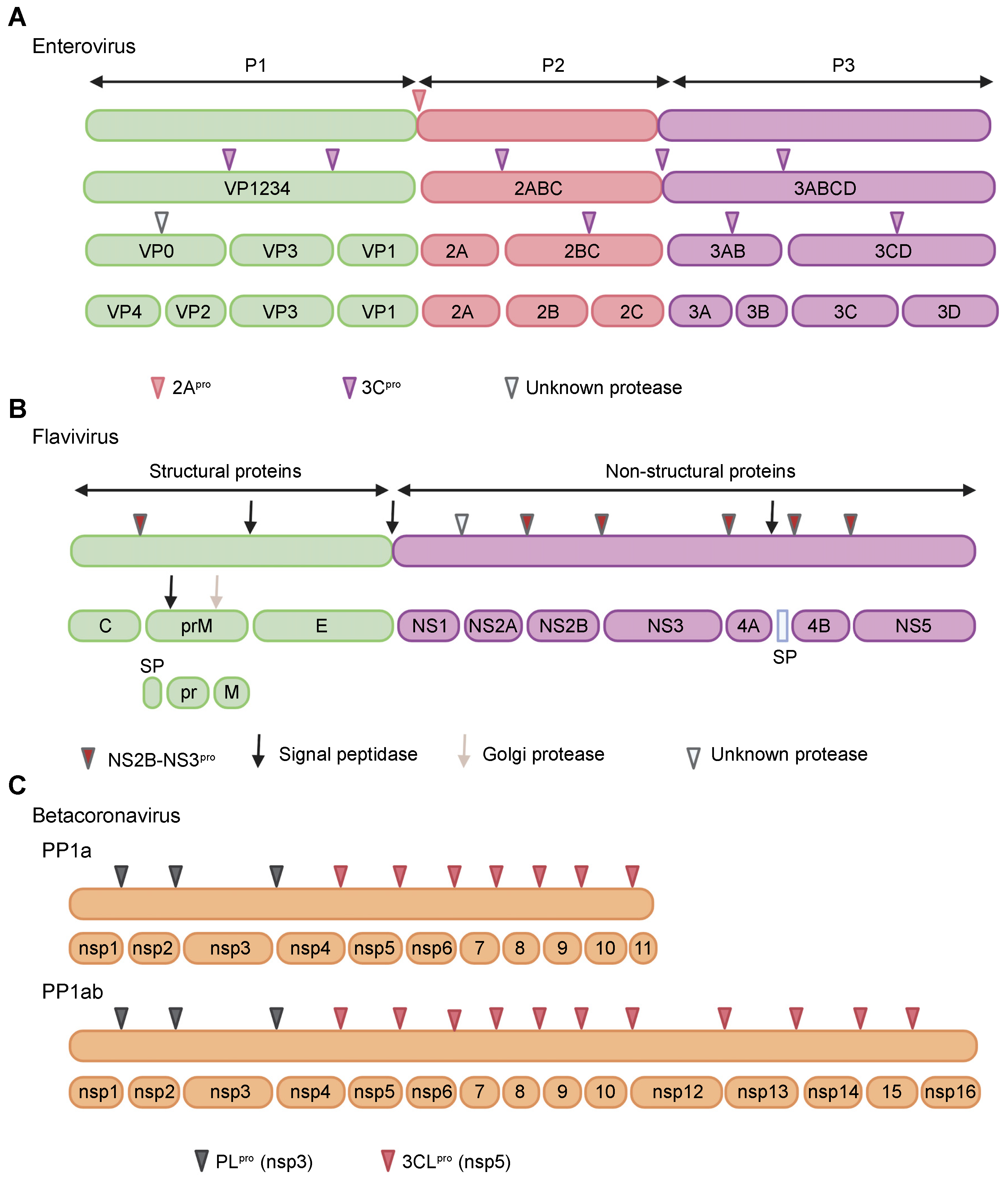
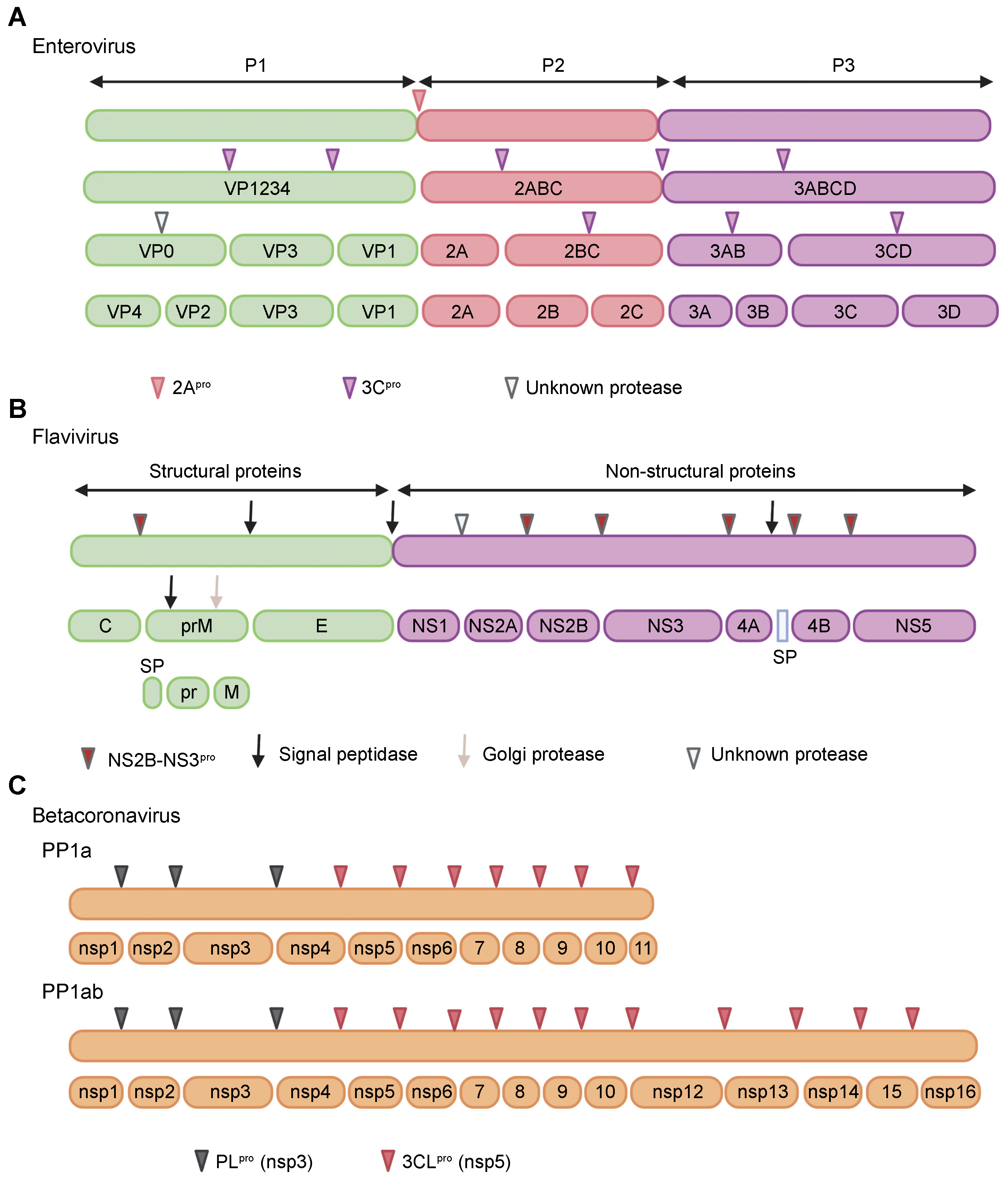
2. Proteases of Positive-Strand RNA Viruses
3. Role of Viral Proteases in Innate Immune Antagonism
3.1. Immune Sensors
3.1.1. Overview
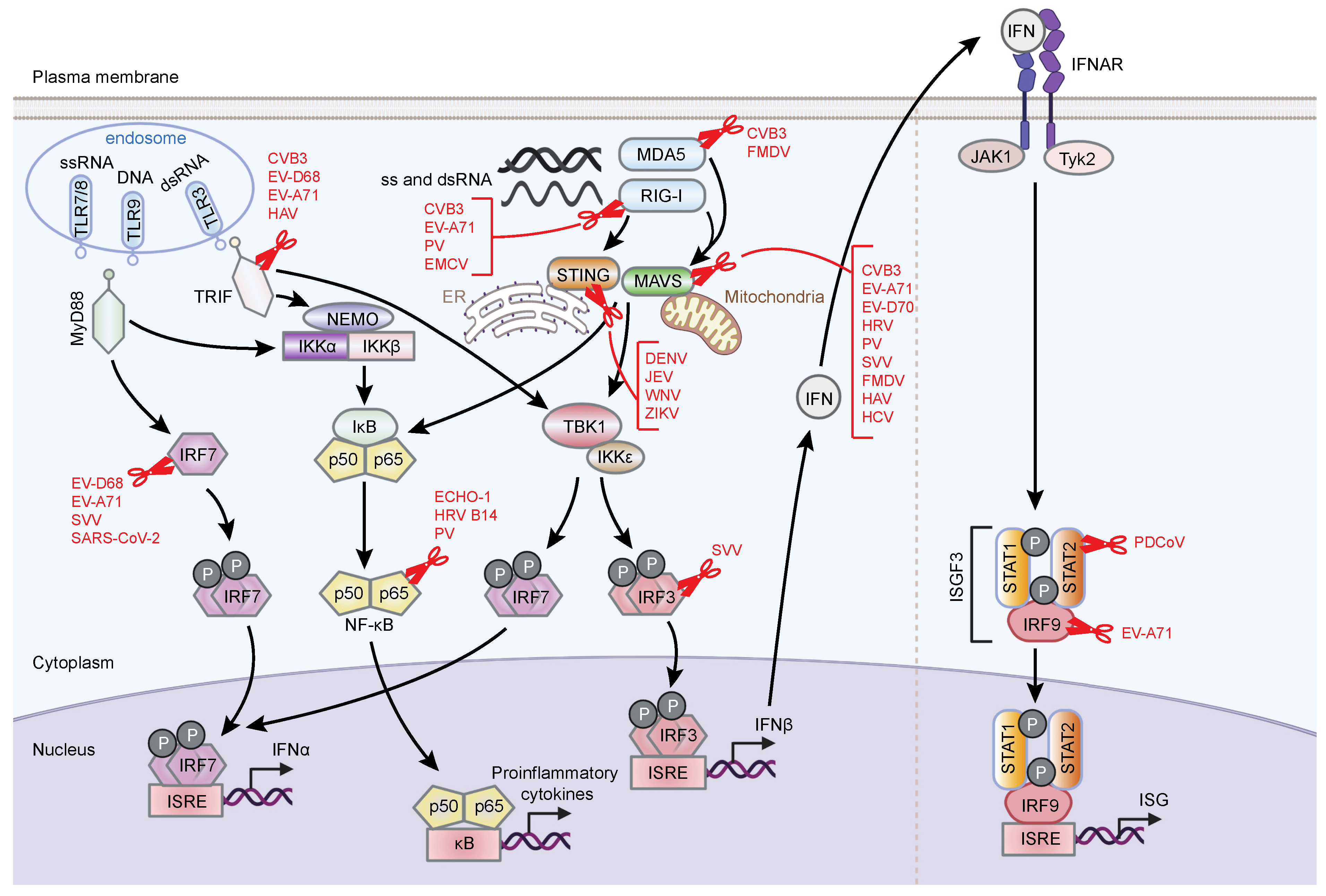
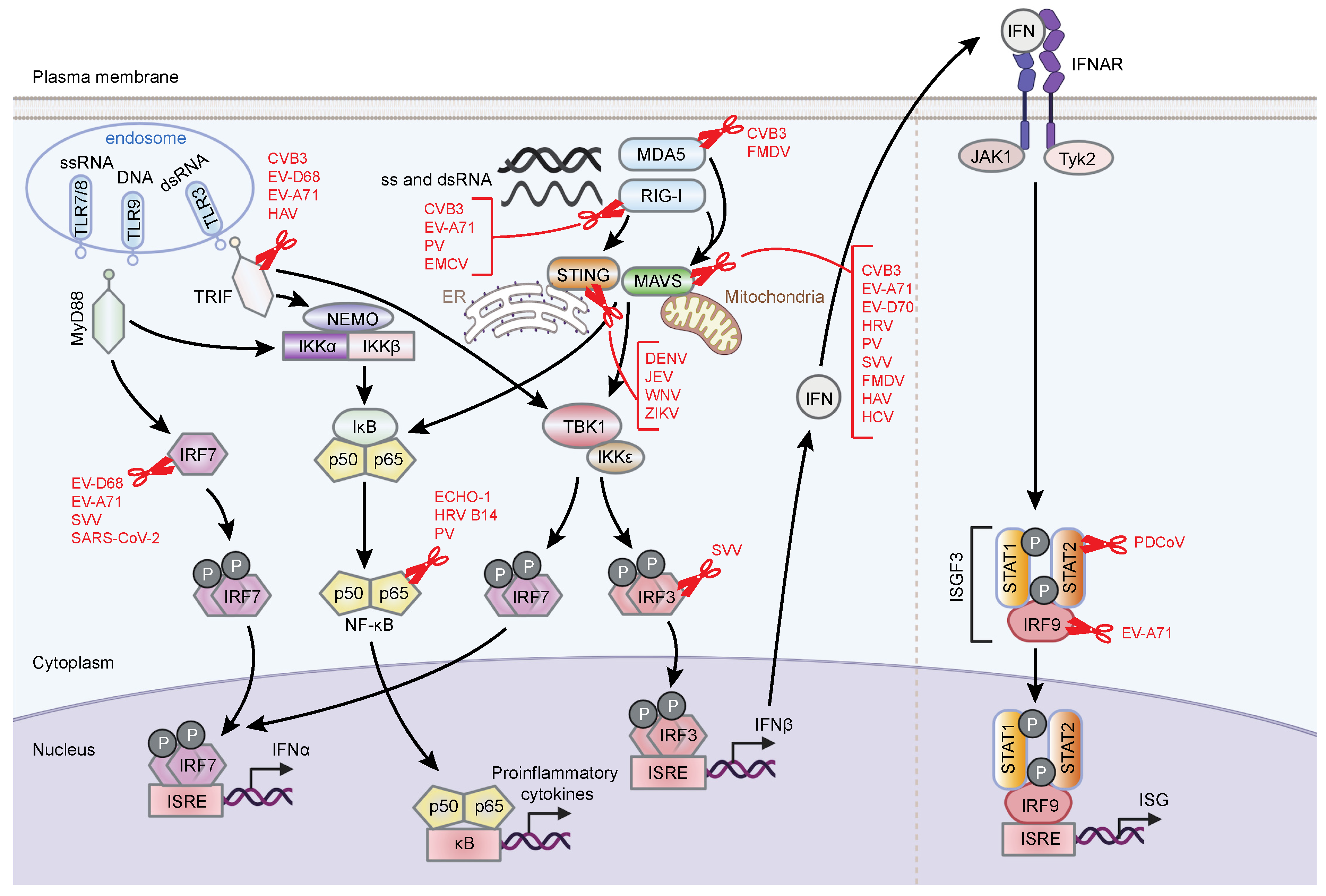
3.1.2. Viral Cleavage of Immune Sensors
3.2. Adaptor Proteins
3.2.1. Overview
3.2.2. Viral Cleavage of Adaptor Proteins
3.3. Transcription Factors
3.3.1. Overview
3.3.2. Viral Cleavage of Transcription Factors
3.4. The IFN Response Pathway
3.4.1. Overview
3.4.2. Ablation of IFN Responses by Viral Proteases
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, H.-H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate Immune Recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr. Approaching the Asymptote? Evolution and Revolution in Immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Katze, M.G.; He, Y.; Gale, M., Jr. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 2016, 17, 523–530. [Google Scholar] [CrossRef]
- Lei, J.; Hilgenfeld, R. RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [Google Scholar] [CrossRef] [Green Version]
- Stanifer, M.L.; Kischnick, C.; Rippert, A.; Albrecht, D.; Boulant, S. Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci. Rep. 2017, 7, 10873. [Google Scholar] [CrossRef]
- Mc Inerney, G.; Hedestam, G.K. Direct Cleavage, Proteasomal Degradation and Sequestration: Three Mechanisms of Viral Subversion of Type I Interferon Responses. J. Innate Immun. 2009, 1, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-Y.; Khan, N.; Close, B.J.; Goel, R.K.; Blum, B.; Tavares, A.H.; Kenney, D.; Conway, H.L.; Ewoldt, J.K.; Chitalia, V.C.; et al. SARS-CoV-2 Disrupts Proximal Elements in the JAK-STAT Pathway. J. Virol. 2021, 95, JVI0086221. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Kapell, S.; Hertz, N.T.; Wu, X.; Bell, K.; Ashbrook, A.W.; Mark, M.T.; Zebroski, H.A.; Neal, M.L.; Flodström-Tullberg, M.; et al. Defining the proteolytic landscape during enterovirus infection. PLOS Pathog. 2020, 16, e1008927. [Google Scholar] [CrossRef]
- Epperson, M.L.; Lee, C.A.; Fremont, D.H. Subversion of cytokine networks by virally encoded decoy receptors. Immunol. Rev. 2012, 250, 199–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, B.E.; Herzog, R.W. Covert Warfare against the Immune System: Decoy Capsids, Stealth Genomes, and Suppressors. Mol. Ther. 2013, 21, 1648–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernaez, B.; Alonso-Lobo, J.M.; Montanuy, I.; Fischer, C.; Sauer, S.; Sigal, L.; Sevilla, N.; Alcamí, A. A virus-encoded type I interferon decoy receptor enables evasion of host immunity through cell-surface binding. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Randall, G. (+) RNA virus replication compartments: A safe home for (most) viral replication. Curr. Opin. Microbiol. 2016, 32, 82–88. [Google Scholar] [CrossRef]
- Wang, Z.W.; Sarmento, L.; Wang, Y.; Li, X.-Q.; Dhingra, V.; Tseggai, T.; Jiang, B.; Fu, Z.F. Attenuated Rabies Virus Activates, while Pathogenic Rabies Virus Evades, the Host Innate Immune Responses in the Central Nervous System. J. Virol. 2005, 79, 12554–12565. [Google Scholar] [CrossRef] [Green Version]
- Langland, J.O.; Kash, J.C.; Carter, V.; Thomas, M.J.; Katze, M.G.; Jacobs, B.L. Suppression of Proinflammatory Signal Transduction and Gene Expression by the DualNucleic Acid Binding Domains of the Vaccinia Virus E3L Proteins. J. Virol. 2006, 80, 10083–10095. [Google Scholar] [CrossRef] [Green Version]
- Kash, J.C.; Muhlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.-D.; Katze, M.G. Global Suppression of the Host Antiviral Response by Ebola- and Marburgviruses: Increased Antagonism of the Type I Interferon Response Is Associated with Enhanced Virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katze, M.G.; Fornek, J.L.; Palermo, R.E.; Walters, K.-A.; Korth, M.J. Innate immune modulation by RNA viruses: Emerging insights from functional genomics. Nat. Rev. Immunol. 2008, 8, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Peng, J.; Yang, W.; Zhu, G.; Ren, J.; Li, D.; Zheng, H. Picornavirus 3C – a protease ensuring virus replication and subverting host responses. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef]
- Ng, C.S.; Stobart, C.C.; Luo, H. Innate immune evasion mediated by picornaviral 3C protease: Possible lessons for coronaviral 3C-like protease? Rev. Med. Virol. 2021, 31, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tsu, B.V.; Fay, E.J.; Nguyen, K.T.; Corley, M.R.; Hosuru, B.; Dominguez, V.A.; Daugherty, M.D. Running with Scissors: Evolutionary Conflicts Between Viral Proteases and the Host Immune System. Front. Immunol. 2021, 12, 769543. [Google Scholar] [CrossRef] [PubMed]
- Kaukinen, P.; Sillanpää, M.; Nousiainen, L.; Melén, K.; Julkunen, I. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKε and TBK1 functions. J. Med. Virol. 2012, 85, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Angleró-Rodríguez, Y.I.; Pantoja, P.; Sariol, C.A. Dengue Virus Subverts the Interferon Induction Pathway via NS2B/3 Protease-IκB Kinase ε Interaction. Clin. Vaccine Immunol. 2013, 21, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.; Markham, R.; et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91, e00546-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Lee, J.-H.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; van Gent, M.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 575, 210–216. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating Activity of SARS-CoV Papain-Like Protease. PLOS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Fang, L.; Li, P.; Sun, L.; Fan, J.; Zhang, Q.; Luo, R.; Liu, X.; Li, K.; Chen, H.; et al. The Leader Proteinase of Foot-and-Mouth Disease Virus Negatively Regulates the Type I Interferon Pathway by Acting as a Viral Deubiquitinase. J. Virol. 2011, 85, 3758–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2020, 84, 4619–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zell, R.; Knowles, N.J.; Simmonds, P. A proposed division of the family Picornaviridae into subfamilies based on phylogenetic relationships and functional genomic organization. Arch. Virol. 2021, 166, 2927–2935. [Google Scholar] [CrossRef]
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 43–89. [Google Scholar] [CrossRef]
- Jiang, P.; Liu, Y.; Ma, H.-C.; Paul, A.V.; Wimmer, E. Picornavirus Morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437. [Google Scholar] [CrossRef] [Green Version]
- Norder, H.; De Palma, A.M.; Selisko, B.; Costenaro, L.; Papageorgiou, N.; Arnan, C.; Coutard, B.; Lantez, V.; De Lamballerie, X.; Baronti, C.; et al. Picornavirus non-structural proteins as targets for new anti-virals with broad activity. Antivir. Res. 2011, 89, 204–218. [Google Scholar] [CrossRef]
- Schutheiss, T.; Kusov, Y.Y.; Gauss-Müller, V. Proteinase 3C of Hepatitis A Virus (HAV) Cleaves the HAV Polyprotein P2-P3 at All Sites Including VP1/2A and 2A/2B. Virology 1994, 198, 275–281. [Google Scholar] [CrossRef]
- Hall, D.J.; Palmenberg, A.C. Cleavage site mutations in the encephalomyocarditis virus P3 region lethally abrogate the normal processing cascade. J. Virol. 1996, 70, 5954–5961. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Cheng, A.; Wang, M.; Jia, R.; Sun, K.; Pan, K.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; et al. Structures and Corresponding Functions of Five Types of Picornaviral 2A Proteins. Front. Microbiol. 2017, 8, 1373. [Google Scholar] [CrossRef] [Green Version]
- Petersen, J.F.; Cherney, M.M.; Liebig, H.D.; Skern, T.; Kuechler, E.; James, M.N. The structure of the 2A proteinase from a common cold virus: A proteinase responsible for the shut-off of host-cell protein synthesis. EMBO J. 1999, 18, 5463–5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, D.A.; Smith, W.W.; Ferre, R.A.; Condon, B.; Budahazi, G.; Slsson, W.; Villafranca, J.; Janson, C.A.; McElroy, H.; Gribskov, C.; et al. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 1994, 77, 761–771. [Google Scholar] [CrossRef]
- Tan, J.; George, S.; Kusov, Y.; Perbandt, M.; Anemüller, S.; Mesters, J.R.; Norder, H.; Coutard, B.; Lacroix, C.; Leyssen, P.; et al. 3C Protease of Enterovirus 68: Structure-Based Design of Michael Acceptor Inhibitors and Their Broad-Spectrum Antiviral Effects against Picornaviruses. J. Virol. 2013, 87, 4339–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipelt, J.; Guarné, A.; Bergmann, E.; James, M.; Sommergruber, W.; Fita, I.; Skern, T. The structures of picornaviral proteinases. Virus Res. 1999, 62, 159–168. [Google Scholar] [CrossRef]
- Guarné, A.; Tormo, J.; Kirchweger, R.; Pfistermueller, D.; Fita, I.; Skern, T. Structure of the foot-and-mouth disease virus leader protease: A papain-like fold adapted for self-processing and eIF4G recognition. EMBO J. 1998, 17, 7469–7479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogle, J.M.; Chow, M.; Filman, D.J. Three-Dimensional Structure of Poliovirus at 2.9 Å Resolution. Science 1985, 229, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar] [CrossRef]
- Falgout, B.; Pethel, M.; Zhang, Y.M.; Lai, C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991, 65, 2467–2475. [Google Scholar] [CrossRef] [Green Version]
- Chambers, T.J.; McCourt, D.W.; Rice, C.M. Production of yellow fever virus proteins in infected cells: Identification of discrete polyprotein species and analysis of cleavage kinetics using region-specific polyclonal antisera. Virology 1990, 177, 159–174. [Google Scholar] [CrossRef]
- Clum, S.; Ebner, K.E.; Padmanabhan, R. Cotranslational Membrane Insertion of the Serine Proteinase Precursor NS2B-NS3(Pro) of Dengue Virus Type 2 Is Required for Efficient in Vitro Processing and Is Mediated through the Hydrophobic Regions of NS2B. J. Biol. Chem. 1997, 272, 30715–30723. [Google Scholar] [CrossRef] [Green Version]
- Jan, L.-R.; Yang, C.-S.; Trent, D.W.; Falgout, B.; Lai, C.-J. Processing of Japanese encephalitis virus non-structural proteins: NS2B-NS3 complex and heterologous proteases. J. Gen. Virol. 1995, 76, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Sampath, A.; Chao, A.; Wen, D.; Nanao, M.; Chene, P.; Vasudevan, S.G.; Lescar, J. Structure of the Dengue Virus Helicase/Nucleoside Triphosphatase Catalytic Domain at a Resolution of 2.4 Å. J. Virol. 2005, 79, 10278–10288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.M.; Gayen, S.; Kang, C.; Joy, J.K.; Huang, Q.; Chen, A.S.; Wee, J.L.K.; Ang, M.J.Y.; Lim, H.A.; Hung, A.W.; et al. NMR Analysis of a Novel Enzymatically Active Unlinked Dengue NS2B-NS3 Protease Complex. J. Biol. Chem. 2013, 288, 12891–12900. [Google Scholar] [CrossRef] [Green Version]
- Phong, W.Y.; Moreland, N.J.; Lim, S.P.; Wen, D.; Paradkar, P.N.; Vasudevan, S.G. Dengue protease activity: The structural integrity and interaction of NS2B with NS3 protease and its potential as a drug target. Biosci. Rep. 2011, 31, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-F.; Wang, S.-H.; Sun, C.-M.; Hu, S.-T.; Syu, W.-J. Activation of dengue protease autocleavage at the NS2B–NS3 junction by recombinant NS3 and GST–NS2B fusion proteins. J. Virol. Methods 2003, 114, 45–54. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Yao, N.; Reichert, P.; Taremi, S.S.; Prosise, W.W.; Weber, P.C. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease–helicase. Structure 1999, 7, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Reed, K.E.; Rice, C.M. Overview of Hepatitis C Virus Genome Structure, Polyprotein Processing, and Protein Properties. Curr. Top. Microbiol. Immuno. 2000, 242, 55–84. [Google Scholar] [CrossRef]
- Barbato, G.; Cicero, D.O.; Nardi, M.; Steinkühler, C.; Cortese, R.; De Francesco, R.; Bazzo, R. The solution structure of the N-terminal proteinase domain of the hepatitis C virus (HCV) NS3 protein provides new insights into its activation and catalytic mechanism. J. Mol. Biol. 1999, 289, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Briggs, J.M. Mechanistic role of NS4A and substrate in the activation of HCV NS3 protease. Proteins Struct. Funct. Bioinform. 2011, 79, 2428–2443. [Google Scholar] [CrossRef]
- Kim, J.; Morgenstern, K.; Lin, C.; Fox, T.; Dwyer, M.; Landro, J.; Chambers, S.; Markland, W.; Lepre, C.; O’Malley, E.; et al. Crystal Structure of the Hepatitis C Virus NS3 Protease Domain Complexed with a Synthetic NS4A Cofactor Peptide. Cell 1996, 87, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Singh, R.; Kaur, J.; Pandey, S.; Sharma, V.; Thakur, L.; Sati, S.; Mani, S.; Asthana, S.; Sharma, T.K.; et al. Wuhan to World: The COVID-19 Pandemic. Front. Cell. Infect. Microbiol. 2021, 11, 596201. [Google Scholar] [CrossRef]
- John Hopkins University of Medicine, Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 22 April 2022).
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Park, S.; Lee, Y.; Michelow, I.C.; Choe, Y.J. Global seasonality of human coronaviruses: A systematic review. Open Forum Infect. Dis. 2020, 7, ofaa443. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Plant, E.P.; Pérez-Alvarado, G.C.; Jacobs, J.L.; Mukhopadhyay, B.; Hennig, M.; Dinman, J.D. A Three-Stemmed mRNA Pseudoknot in the SARS Coronavirus Frameshift Signal. PLoS Biol. 2005, 3, e172. [Google Scholar] [CrossRef] [Green Version]
- Brierley, I.; Digard, P.; Inglis, S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal: Requirement for an RNA pseudoknot. Cell 1989, 57, 537–547. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Siddell, S.G. Processing of the Human Coronavirus 229E Replicase Polyproteins by the Virus-Encoded 3C-Like Proteinase: Identification of Proteolytic Products and Cleavage Sites Common to pp1a and pp1ab. J. Virol. 1999, 73, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Hilgenfeld, R. Structural and mutational analysis of the interaction between the Middle-East respiratory syndrome coronavirus (MERS-CoV) papain-like protease and human ubiquitin. Virol. Sin. 2016, 31, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Mesters, J.R.; Drosten, C.; Anemüller, S.; Ma, Q.; Hilgenfeld, R. Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antivir. Res. 2014, 109, 72–82. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative Expression of Toll-Like Receptor 1–10 mRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Ezhong, Y.; Ekinio, A.; Esaleh, M. Functions of NOD-Like Receptors in Human Diseases. Front. Immunol. 2013, 4, 333. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Kang, D.-C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. 2002, 99, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Rothenfusser, S.; Goutagny, N.; DiPerna, G.; Gong, M.; Monks, B.G.; Schoenemeyer, A.; Yamamoto, M.; Akira, S.; Fitzgerald, K.A. The RNA Helicase Lgp2 Inhibits TLR-Independent Sensing of Viral Replication by Retinoic Acid-Inducible Gene-I. J. Immunol. 2005, 175, 5260–5268. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.H.; Reimer, T.; Kim, Y.-G.; Nuñez, G. NOD-like receptors (NLRs): Bona fide intracellular microbial sensors. Curr. Opin. Immunol. 2008, 20, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Inohara, N.; Nuñez, G. The NOD: A signaling module that regulates apoptosis and host defense against pathogens. Oncogene 2001, 20, 6473–6481. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2A pro Targets MDA5 and MAVS in Infected Cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulido, M.R.; Martínez-Salas, E.; Sobrino, F.; Sáiz, M. MDA5 cleavage by the Leader protease of foot-and-mouth disease virus reveals its pleiotropic effect against the host antiviral response. Cell Death Dis. 2020, 11, 718. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C Protein of Enterovirus 71 Inhibits Retinoid Acid-Inducible Gene I-Mediated Interferon Regulatory Factor 3 Activation and Type I Interferon Responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef] [Green Version]
- Papon, L.; Oteiza, A.; Imaizumi, T.; Kato, H.; Brocchi, E.; Lawson, T.G.; Akira, S.; Mechti, N. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 2009, 393, 311–318. [Google Scholar] [CrossRef]
- Qu, L.; Feng, Z.; Yamane, D.; Liang, Y.; Lanford, R.E.; Li, K.; Lemon, S.M. Disruption of TLR3 Signaling Due to Cleavage of TRIF by the Hepatitis A Virus Protease-Polymerase Processing Intermediate, 3CD. PLoS Pathog. 2011, 7, e1002169. [Google Scholar] [CrossRef]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef]
- Warner, N.; Núñez, G. MyD88: A Critical Adaptor Protein in Innate Immunity Signal Transduction. J. Immunol. 2012, 190, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptor and RIG-1-like Receptor Signaling. Ann. N. Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Häcker, H.; Karin, M. Regulation and Function of IKK and IKK-Related Kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting Edge: A Novel Toll/IL-1 Receptor Domain-Containing Adapter That Preferentially Activates the IFN-β Promoter in the Toll-Like Receptor Signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Takeuchi, O.; Fujita, T.; Inoue, J.-I.; Mühlradt, P.F.; Sato, S.; Hoshino, K.; Akira, S. Lipopolysaccharide Stimulates the MyD88-Independent Pathway and Results in Activation of IFN-Regulatory Factor 3 and the Expression of a Subset of Lipopolysaccharide-Inducible Genes. J. Immunol. 2001, 167, 5887–5894. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.-Y.; Han, K.-J.; Li, L.-Y.; Zhai, Z.; Shu, H.-B. VISA Is an Adapter Protein Required for Virus-Triggered IFN-β Signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.-X.; Halfmann, R.; Chen, Z.J. Prion-like Polymerization Underlies Signal Transduction in Antiviral Immune Defense and Inflammasome Activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, E.D.; Wang, C.-Y. MAVS Self-Association Mediates Antiviral Innate Immune Signaling. J. Virol. 2009, 83, 3420–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.; Whelan, S.; Fransen, M.; et al. Peroxisomes Are Signaling Platforms for Antiviral Innate Immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, J.M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2008, 227, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Genin, P.; Civas, A.; Hiscott, J. RIG-I-like receptors: Sensing and responding to RNA virus infection. Semin. Immunol. 2009, 21, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653–8658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Waterman, P.M.; Jonscher, K.R.; Short, C.M.; Reisdorph, N.A.; Cambier, J.C. MPYS, a Novel Membrane Tetraspanner, Is Associated with Major Histocompatibility Complex Class II and Mediates Transduction of Apoptotic Signals. Mol. Cell. Biol. 2008, 28, 5014–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazmi, A.; Mukhopadhyay, R.; Dutta, K.; Basu, A. STING Mediates Neuronal Innate Immune Response Following Japanese Encephalitis Virus Infection. Sci. Rep. 2012, 2, 347. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Dai, T.; He, X.; Zhang, Z.; Xie, F.; Wang, S.; Zhang, L.; Zhou, F. The interactions between cGAS-STING pathway and pathogens. Signal Transduct. Target. Ther. 2020, 5, 91. [Google Scholar] [CrossRef]
- Aguirre, S.; Fernandez-Sesma, A. Collateral Damage during Dengue Virus Infection: Making Sense of DNA by cGAS. J. Virol. 2017, 91, e01081-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3Cpro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLOS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 Protease 2Apro Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLOS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [Green Version]
- Lind, K.; Svedin, E.; Domsgen, E.; Kapell, S.; Laitinen, O.; Moll, M.; Tullberg, M.F. Coxsackievirus counters the host innate immune response by blocking type III interferon expression. J. Gen. Virol. 2016, 97, 1368–1380. [Google Scholar] [CrossRef]
- Qian, S.; Fan, W.; Liu, T.; Wu, M.; Zhang, H.; Cui, X.; Zhou, Y.; Hu, J.; Wei, S.; Chen, H.; et al. Seneca Valley Virus Suppresses Host Type I Interferon Production by Targeting Adaptor Proteins MAVS, TRIF, and TANK for Cleavage. J. Virol. 2017, 91, e00823-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, L.J.; Aloise, C.; Swatek, K.N.; Medina, G.N.; Olek, K.M.; Rabouw, H.H.; De Groot, R.J.; Langereis, M.A.; Santos, T.D.L.; Komander, D.; et al. Dissecting distinct proteolytic activities of FMDV Lpro implicates cleavage and degradation of RLR signaling proteins, not its deISGylase/DUB activity, in type I interferon suppression. PLoS Pathog. 2020, 16, e1008702. [Google Scholar] [CrossRef] [PubMed]
- Bellecave, P.; Filipowicz, M.; Donzé, O.; Kennel, A.; Gouttenoire, J.; Meylan, E.; Terracciano, L.; Tschopp, J.; Sarrazin, C.; Berg, T.; et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology 2009, 51, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Lacoste, J.; Nakhaei, P.; Sun, Q.; Yang, L.; Paz, S.; Wilkinson, P.; Julkunen, I.; Vitour, D.; Meurs, E.; et al. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKε Molecular Complex from the Mitochondrial Outer Membrane by Hepatitis C Virus NS3-4A Proteolytic Cleavage. J. Virol. 2006, 80, 6072–6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Li, L.; Lei, X.; Zhou, H.; Zhou, Z.; He, B.; Wang, J. Enterovirus 68 3C Protease Cleaves TRIF To Attenuate Antiviral Responses Mediated by Toll-Like Receptor 3. J. Virol. 2014, 88, 6650–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the Adaptor Protein TRIF by Enterovirus 71 3C Inhibits Antiviral Responses Mediated by Toll-Like Receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; PLoss, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. USA 2018, 115, E6310–E6318. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. Dengue Virus Targets the Adaptor Protein MITA to Subvert Host Innate Immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef]
- Stabell, A.C.; Meyerson, N.R.; Gullberg, R.C.; Gilchrist, A.R.; Webb, K.J.; Old, W.M.; Perera, R.; Sawyer, S.L. Dengue viruses cleave STING in humans but not in nonhuman primates, their presumed natural reservoir. eLife 2018, 7, e31919. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, L.M. The Role of Nuclear Factor κB in the Interferon Response. J. Interf. Cytokine Res. 2011, 31, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, H.; Negishi, H.; Taniguchi, T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. OncoImmunology 2012, 1, 1376–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF- B Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Sun, S.-C.; Chang, J.-H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Delhase, M. The IκB kinase (IKK) and NF-κB: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Sun, S.-C.; Ley, S.C. New insights into NF-κB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Beinke, S.; Ley, S.C. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.E.; Huang, D.-B.; Chen, Y.-Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Sakakibara, J.; Sudo, Y.; Miyamoto, M.; Kimura, Y.; Taniguchi, T. Evidence for a nuclear factor(s), IRF-1, mediating induction and silencing properties to human IFN-beta gene regulatory elements. EMBO J. 1988, 7, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Fujita, T.; Kimura, Y.; Maruyama, M.; Harada, H.; Sudo, Y.; Miyata, T.; Taniguchi, T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-β gene regulatory elements. Cell 1988, 54, 903–913. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFκB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Response to Viruses for IFN-α/β Gene Induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Panne, D.; Maniatis, T.; Harrison, S.C. An Atomic Model of the Interferon-β Enhanceosome. Cell 2007, 129, 1111–1123. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Mamane, Y.; Hiscott, J. Structural and Functional Analysis of Interferon Regulatory Factor 3: Localization of the Transactivation and Autoinhibitory Domains. Mol. Cell. Biol. 1999, 19, 2465–2474. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Mamane, Y.; Hiscott, J. Multiple Regulatory Domains Control IRF-7 Activity in Response to Virus Infection. J. Biol. Chem. 2000, 275, 34320–34327. [Google Scholar] [CrossRef] [Green Version]
- Tenoever, B.R.; Sharma, S.; Zou, W.; Sun, Q.; Grandvaux, N.; Julkunen, I.; Hemmi, H.; Yamamoto, M.; Akira, S.; Yeh, W.-C.; et al. Activation of TBK1 and IKKε Kinases by Vesicular Stomatitis Virus Infection and the Role of Viral Ribonucleoprotein in the Development of Interferon Antiviral Immunity. J. Virol. 2004, 78, 10636–10649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farlik, M.; Rapp, B.; Marie, I.; Levy, D.E.; Jamieson, A.M.; Decker, T. Contribution of a TANK-Binding Kinase 1–Interferon (IFN) Regulatory Factor 7 Pathway to IFN-γ-Induced Gene Expression. Mol. Cell. Biol. 2012, 32, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, K.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Sharma, S.; Tenoever, B.R.; Grandvaux, N.; Zhou, G.-P.; Lin, R.; Hiscott, J. Triggering the Interferon Antiviral Response Through an IKK-Related Pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.; Lancaster, A.; Wilkins, C.; Suthar, M.S.; Huang, A.; Vick, S.C.; Clepper, L.; Thackray, L.; Brassil, M.M.; Virgin, H.; et al. IRF-3, IRF-5, and IRF-7 Coordinately Regulate the Type I IFN Response in Myeloid Dendritic Cells Downstream of MAVS Signaling. PLoS Pathog. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of Interferon Regulatory Factor 7 by Enterovirus 71 3C Suppresses Cellular Responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Liu, L.; Lei, X.; Zhou, Z.; He, B.; Wang, J. 3C Protease of Enterovirus D68 Inhibits Cellular Defense Mediated by Interferon Regulatory Factor 7. J. Virol. 2016, 90, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Liu, H.; Zhu, Z.; Yang, F.; Ma, L.; Cai, X.; Xue, Q.; Zheng, H. Seneca Valley Virus 3Cpro abrogates the IRF3- and IRF7-mediated innate immune response by degrading IRF3 and IRF7. Virology 2018, 518, 1–7. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Neznanov, N.; Chumakov, K.M.; Neznanova, L.; Almasan, A.; Banerjee, A.K.; Gudkov, A.V. Proteolytic Cleavage of the p65-RelA Subunit of NF-κB during Poliovirus Infection. J. Biol. Chem. 2005, 280, 24153–24158. [Google Scholar] [CrossRef] [Green Version]
- Gagné, B.; Tremblay, N.; Park, A.Y.; Baril, M.; Lamarre, D. Importin β1 targeting by hepatitis C virus NS3/4A protein restricts IRF3 and NF-κB signaling of IFNB1 antiviral response. Traffic 2017, 18, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, C.; Levy, D.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.M.; Racaniello, V.R. Proteinase 2Apro Is Essential for Enterovirus Replication in Type I Interferon-Treated Cells. J. Virol. 2009, 83, 4412–4422. [Google Scholar] [CrossRef] [Green Version]
- Hung, H.-C.; Wang, H.-C.; Shih, S.-R.; Teng, I.-F.; Tseng, C.-P.; Hsu, J.T.-A. Synergistic Inhibition of Enterovirus 71 Replication by Interferon and Rupintrivir. J. Infect. Dis. 2011, 203, 1784–1790. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Wang, D.; Zhou, J.; Pan, T.; Chen, J.; Yang, Y.; Lv, M.; Ye, X.; Peng, G.; Fang, L.; et al. Porcine Deltacoronavirus nsp5 Antagonizes Type I Interferon Signaling by Cleaving STAT2. J. Virol. 2017, 91, e00003-17. [Google Scholar] [CrossRef] [Green Version]
- Meyer, B.; Chiaravalli, J.; Gellenoncourt, S.; Brownridge, P.; Bryne, D.P.; Daly, L.A.; Grauslys, A.; Walter, M.; Agou, F.; Chakrabarti, L.A.; et al. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021, 12, 5553. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chin, C.V.; Saeed, M. Surgical Strikes on Host Defenses: Role of the Viral Protease Activity in Innate Immune Antagonism. Pathogens 2022, 11, 522. https://doi.org/10.3390/pathogens11050522
Chin CV, Saeed M. Surgical Strikes on Host Defenses: Role of the Viral Protease Activity in Innate Immune Antagonism. Pathogens. 2022; 11(5):522. https://doi.org/10.3390/pathogens11050522
Chicago/Turabian StyleChin, Chue Vin, and Mohsan Saeed. 2022. "Surgical Strikes on Host Defenses: Role of the Viral Protease Activity in Innate Immune Antagonism" Pathogens 11, no. 5: 522. https://doi.org/10.3390/pathogens11050522
APA StyleChin, C. V., & Saeed, M. (2022). Surgical Strikes on Host Defenses: Role of the Viral Protease Activity in Innate Immune Antagonism. Pathogens, 11(5), 522. https://doi.org/10.3390/pathogens11050522