
[PRION+] States Are Associated with Specific Histone H3 Post-Translational Modification Changes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Different [PRION+] States Connect to Unique Histone PTM Alterations
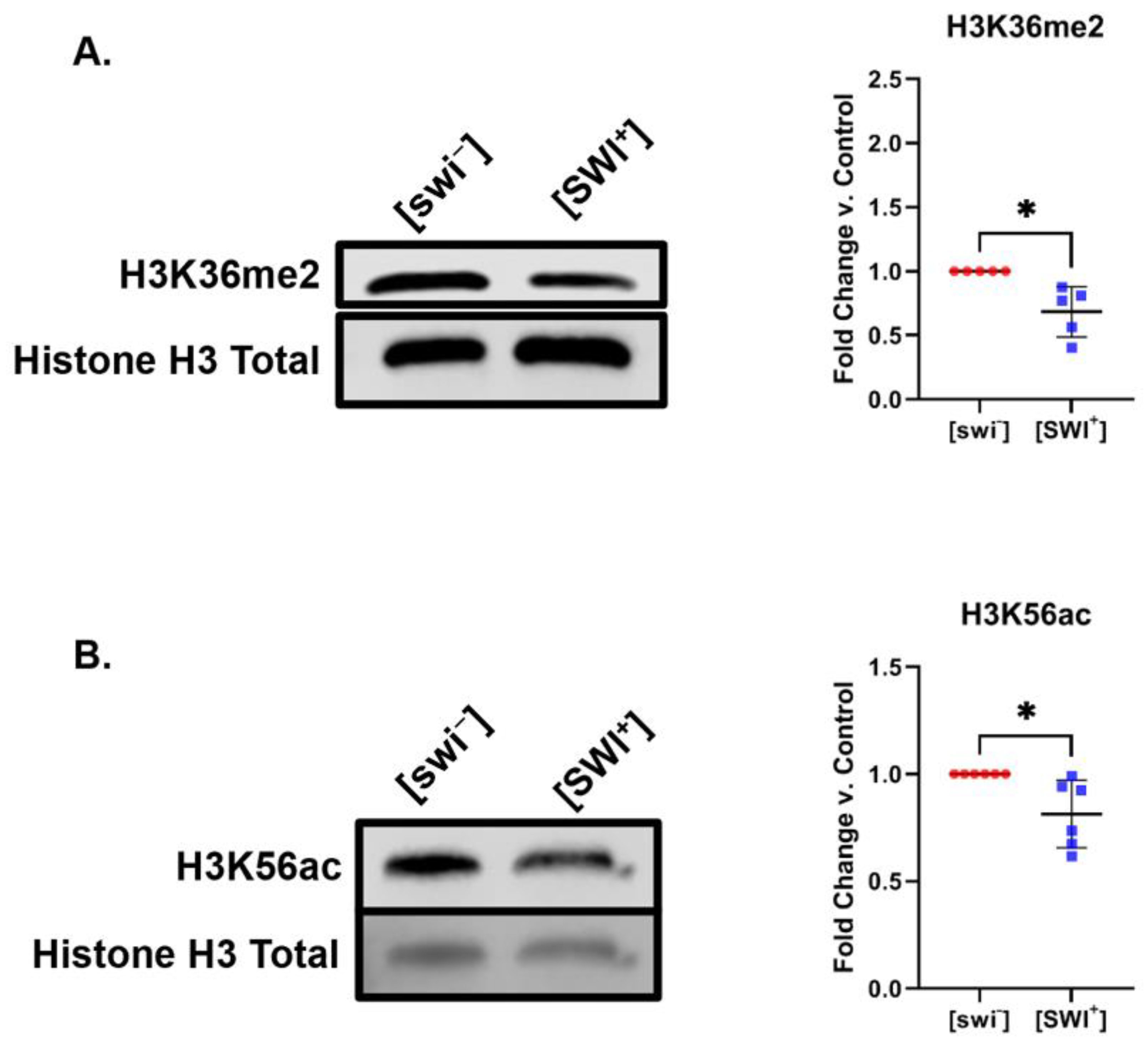
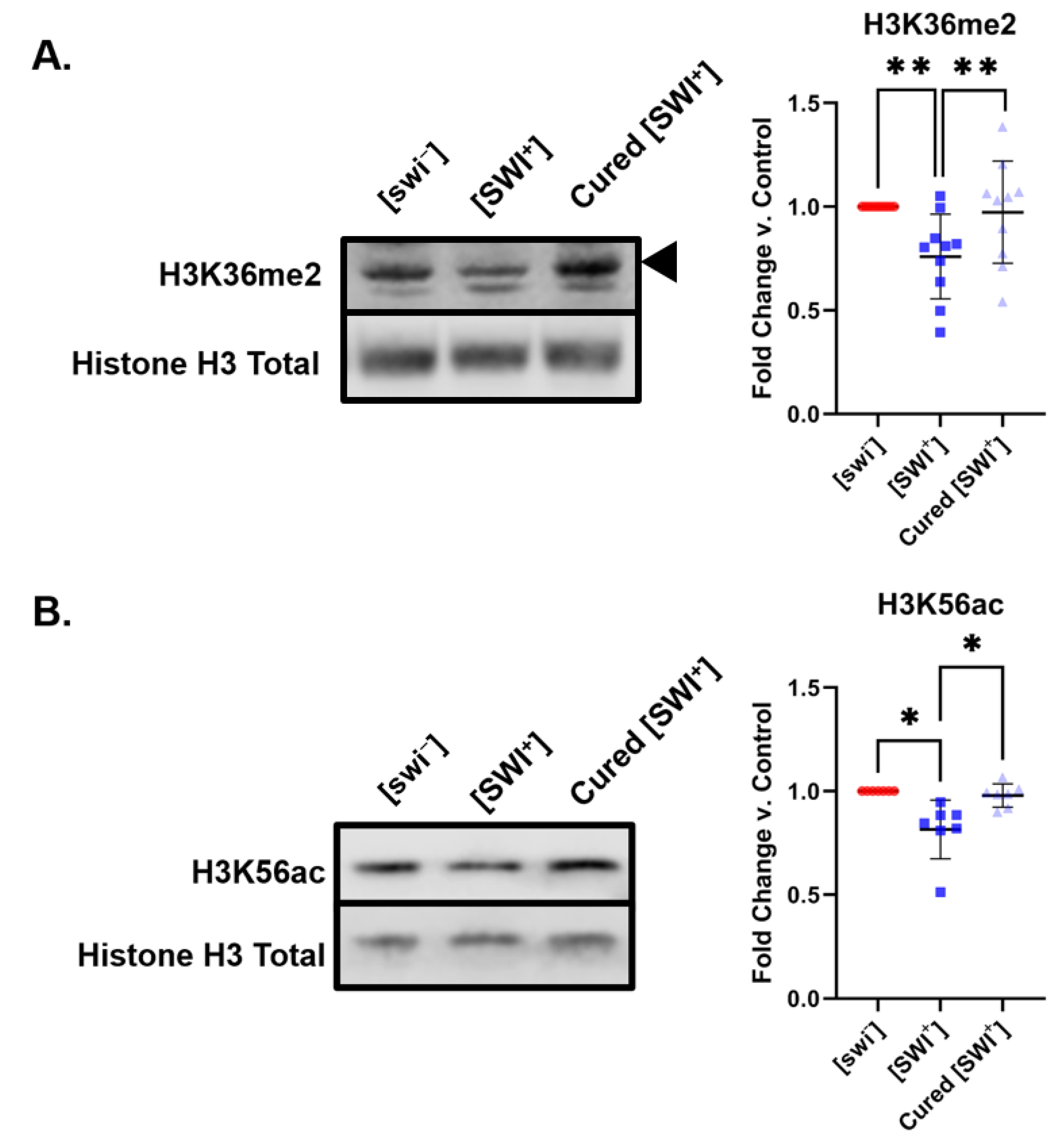
2.2. [SWI+] Is Linked to Decreases in H3K36me2 and H3K56ac Levels
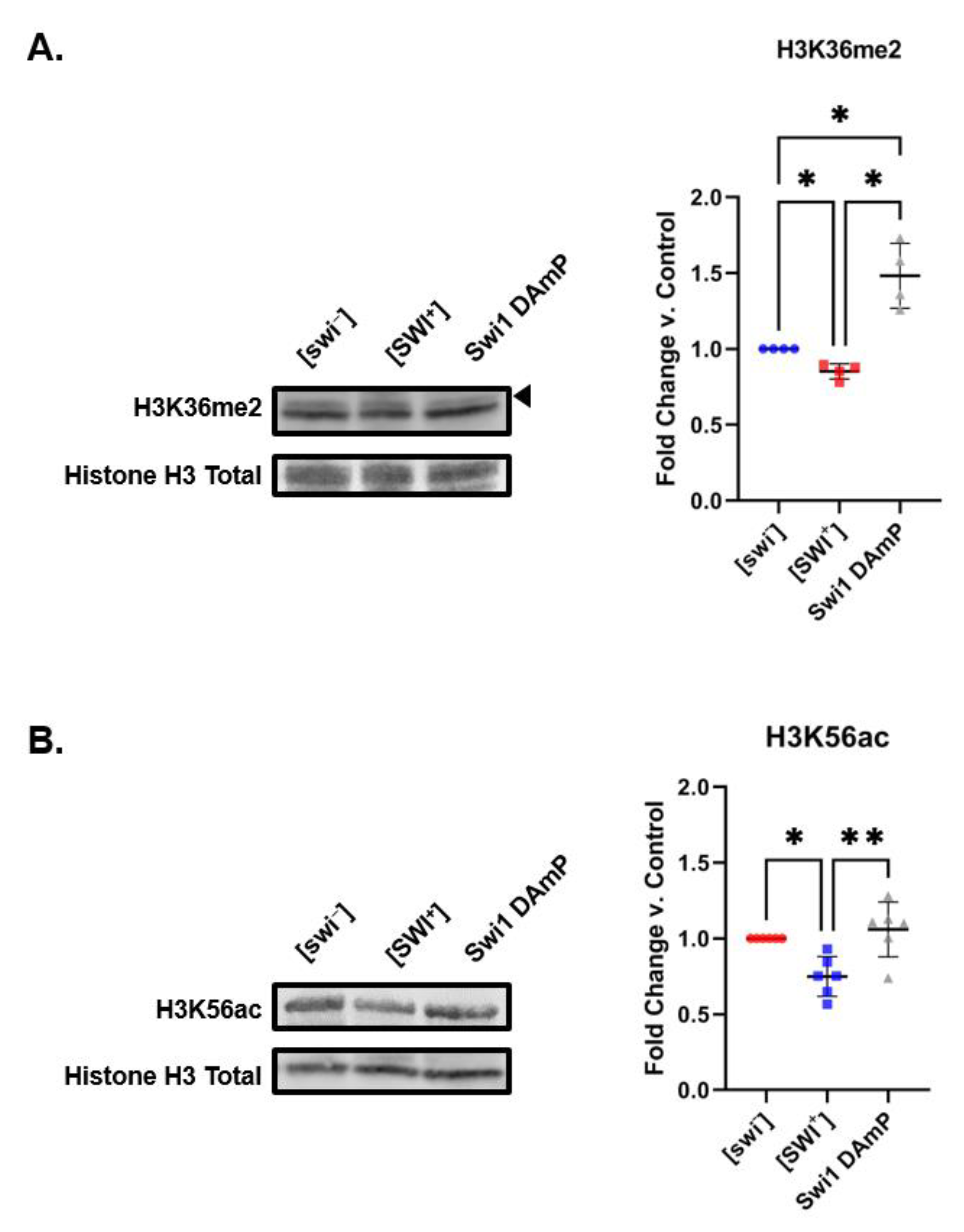
2.3. Reduced Swi1 Expression Is Associated with Different Histone PTM Changes That Differ from Those Observed in [SWI+]
2.4. [PIN+] Is Associated with Decreases in Di- and Trimethylation of Histone H3 at Specific Sites
2.5. Rnq1 Deletion Reveals Histone PTM Changes That Differ from Those Observed in [PIN+]
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Yeast Strains
4.3. Yeast Culture
4.4. Prion Curing
4.5. RNA Purification and Quantification
4.6. Western Blotting
4.7. Filter Retention Assay
4.8. Flocculation Measurements
4.9. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aguzzi, A.; Polymenidou, M. Mammalian prion biology: One century of evolving concepts. Cell 2004, 116, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Lindquist, S. Epigenetics in the extreme: Prions and the inheritance of environmentally acquired traits. Science 2010, 330, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Galopier, A.; Martini, C.; Matsufuji, S.; Fabret, C.; Rousset, J.P. Epigenetic control of polyamines by the prion [PSI+]. Nat. Cell Biol. 2008, 10, 1069–1075. [Google Scholar] [CrossRef]
- Joseph, S.B.; Kirkpatrick, M. Effects of the [PSI+] prion on rates of adaptation in yeast. J. Evol. Biol. 2008, 21, 773–780. [Google Scholar] [CrossRef] [PubMed]
- True, H.L.; Lindquist, S.L. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000, 407, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Eaglestone, S.S.; Cox, B.S.; Tuite, M.F. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J. 1999, 18, 1974–1981. [Google Scholar] [CrossRef] [PubMed]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nuske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359, eaao5654. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Shewmaker, F.P.; Bateman, D.A.; Edskes, H.K.; Gorkovskiy, A.; Dayani, Y.; Bezsonov, E.E. Yeast prions: Structure, biology, and prion-handling systems. Microbiol. Mol. Biol. Rev. 2015, 79, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.; Krobitsch, S.; Li, L.; Sondheimer, N. Investigating protein conformation-based inheritance and disease in yeast. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Goncharoff, D.K.; Du, Z.; Li, L. A brief overview of the Swi1 prion-[SWI+]. FEMS Yeast Res. 2018, 18, foy061. [Google Scholar] [CrossRef] [PubMed]
- Sondheimer, N.; Lindquist, S. Rnq1: An epigenetic modifier of protein function in yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Harvey, Z.H.; Chakravarty, A.K.; Futia, R.A.; Jarosz, D.F. A Prion Epigenetic Switch Establishes an Active Chromatin State. Cell 2020, 180, 928–940.e914. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanam, P.; Winston, F. The Swi/Snf family nucleosome-remodeling complexes and transcriptional control. Trends Genet. 2000, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhang, Y.; Li, L. The Yeast Prion [SWI(+)] Abolishes Multicellular Growth by Triggering Conformational Changes of Multiple Regulators Required for Flocculin Gene Expression. Cell Rep. 2015, 13, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.M.; Regenberg, B. Multicellular group formation in Saccharomyces cerevisiae. Proc. Biol. Sci. 2019, 286, 20191098. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, L.F.; Kennedy, B.K.; Harlow, E. A large-scale overexpression screen in Saccharomyces cerevisiae identifies previously uncharacterized cell cycle genes. Proc. Natl. Acad. Sci. USA 2001, 98, 3946–3951. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, H.; Shibata, S.; Ishiwata, M.; Nakamura, Y. Selfish prion of Rnq1 mutant in yeast. Genes Cells 2009, 14, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Pijnappel, W.W.; Schaft, D.; Roguev, A.; Shevchenko, A.; Tekotte, H.; Wilm, M.; Rigaut, G.; Seraphin, B.; Aasland, R.; Stewart, A.F. The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev. 2001, 15, 2991–3004. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Ntranos, A.; Casaccia, P. Bromodomains: Translating the words of lysine acetylation into myelin injury and repair. Neurosci. Lett. 2016, 625, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Miller, T.; Richon, V.M. Histone deacetylases. Curr. Opin. Pharmacol. 2003, 3, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Regan, J.; Bartom, E.; Wu, W.S.; Zhang, L.; Goncharoff, D.K.; Li, L. Elucidating the regulatory mechanism of Swi1 prion in global transcription and stress responses. Sci. Rep. 2020, 10, 21838. [Google Scholar] [CrossRef]
- Baudin-Baillieu, A.; Legendre, R.; Kuchly, C.; Hatin, I.; Demais, S.; Mestdagh, C.; Gautheret, D.; Namy, O. Genome-wide translational changes induced by the prion [PSI+]. Cell Rep. 2014, 8, 439–448. [Google Scholar] [CrossRef]
- Kundu, S.; Horn, P.J.; Peterson, C.L. SWI/SNF is required for transcriptional memory at the yeast GAL gene cluster. Genes Dev. 2007, 21, 997–1004. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Fnu, S.; Williamson, E.A.; De Haro, L.P.; Brenneman, M.; Wray, J.; Shaheen, M.; Radhakrishnan, K.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2011, 108, 540–545. [Google Scholar] [CrossRef]
- Weinberg, D.N.; Papillon-Cavanagh, S.; Chen, H.; Yue, Y.; Chen, X.; Rajagopalan, K.N.; Horth, C.; McGuire, J.T.; Xu, X.; Nikbakht, H.; et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019, 573, 281–286. [Google Scholar] [CrossRef]
- Li, B.; Jackson, J.; Simon, M.D.; Fleharty, B.; Gogol, M.; Seidel, C.; Workman, J.L.; Shilatifard, A. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 2009, 284, 7970–7976. [Google Scholar] [CrossRef]
- Krogan, N.J.; Kim, M.; Tong, A.; Golshani, A.; Cagney, G.; Canadien, V.; Richards, D.P.; Beattie, B.K.; Emili, A.; Boone, C.; et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell Biol. 2003, 23, 4207–4218. [Google Scholar] [CrossRef]
- Abshiru, N.; Rajan, R.E.; Verreault, A.; Thibault, P. Unraveling Site-Specific and Combinatorial Histone Modifications Using High-Resolution Mass Spectrometry in Histone Deacetylase Mutants of Fission Yeast. J. Proteome Res. 2016, 15, 2132–2142. [Google Scholar] [CrossRef]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef]
- Han, J.; Zhou, H.; Horazdovsky, B.; Zhang, K.; Xu, R.M.; Zhang, Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007, 315, 653–655. [Google Scholar] [CrossRef]
- Kadyrova, L.Y.; Mertz, T.M.; Zhang, Y.; Northam, M.R.; Sheng, Z.; Lobachev, K.S.; Shcherbakova, P.V.; Kadyrov, F.A. A reversible histone H3 acetylation cooperates with mismatch repair and replicative polymerases in maintaining genome stability. PLoS Genet. 2013, 9, e1003899. [Google Scholar] [CrossRef]
- Stejskal, S.; Stepka, K.; Tesarova, L.; Stejskal, K.; Matejkova, M.; Simara, P.; Zdrahal, Z.; Koutna, I. Cell cycle-dependent changes in H3K56ac in human cells. Cell Cycle 2015, 14, 3851–3863. [Google Scholar] [CrossRef]
- Park, Y.N.; Morales, D.; Rubinson, E.H.; Masison, D.; Eisenberg, E.; Greene, L.E. Differences in the curing of [PSI+] prion by various methods of Hsp104 inactivation. PLoS ONE 2012, 7, e37692. [Google Scholar] [CrossRef]
- Ferreira, P.C.; Ness, F.; Edwards, S.R.; Cox, B.S.; Tuite, M.F. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 2001, 40, 1357–1369. [Google Scholar] [CrossRef]
- Romanova, N.V.; Chernoff, Y.O. Hsp104 and prion propagation. Protein Pept. Lett. 2009, 16, 598–605. [Google Scholar] [CrossRef]
- Ness, F.; Ferreira, P.; Cox, B.S.; Tuite, M.F. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol. Cell Biol. 2002, 22, 5593–5605. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Cameron, D.M.; Collins, S.R.; Schuldiner, M.; Stewart-Ornstein, J.; Newman, H.W.; Braun, S.; Madhani, H.D.; Krogan, N.J.; Weissman, J.S. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 2008, 5, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Vitrenko, Y.A.; Pavon, M.E.; Stone, S.I.; Liebman, S.W. Propagation of the [PIN+] prion by fragments of Rnq1 fused to GFP. Curr. Genet. 2007, 51, 309–319. [Google Scholar] [CrossRef][Green Version]
- Wickner, R.B.; Dyda, F.; Tycko, R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register beta-sheet structure. Proc. Natl. Acad. Sci. USA 2008, 105, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Goehler, H.; Droge, A.; Lurz, R.; Schnoegl, S.; Chernoff, Y.O.; Wanker, E.E. Pathogenic polyglutamine tracts are potent inducers of spontaneous Sup35 and Rnq1 amyloidogenesis. PLoS ONE 2010, 5, e9642. [Google Scholar] [CrossRef]
- Sharma, J.; Liebman, S.W. Exploring the basis of [PIN(+)] variant differences in [PSI(+)] induction. J. Mol. Biol. 2013, 425, 3046–3059. [Google Scholar] [CrossRef] [PubMed]
- Topal, S.; Vasseur, P.; Radman-Livaja, M.; Peterson, C.L. Distinct transcriptional roles for Histone H3-K56 acetylation during the cell cycle in Yeast. Nat. Commun. 2019, 10, 4372. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, R.G.; Hooda, J.; Shah, A.; Cao, T.M.; Henke, R.M.; Zhang, L. The nuclear localization of SWI/SNF proteins is subjected to oxygen regulation. Cell Biosci. 2012, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Park, K.W.; Yu, H.; Fan, Q.; Li, L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 2008, 40, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Wickner, R.B.; Shewmaker, F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2011, 2, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, M.; Tradewell, M.L.; Mattina, K.R.; Minotti, S.; Yang, W.; Zhou, H.; Strong, M.J.; Hayward, L.J.; Durham, H.D. Cytoplasmic sequestration of FUS/TLS associated with ALS alters histone marks through loss of nuclear protein arginine methyltransferase 1. Hum. Mol. Genet. 2015, 24, 773–786. [Google Scholar] [CrossRef]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L.; et al. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, S.P.; Barki, L.; Neves-Costa, A.; Santos, F.; Dean, W.; Hawkes, N.; Choudhary, P.; Will, W.R.; Webster, J.; Oxley, D.; et al. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 2011, 42, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Du, H.N.; Fingerman, I.M.; Briggs, S.D. Histone H3 K36 methylation is mediated by a trans-histone methylation pathway involving an interaction between Set2 and histone H4. Genes Dev. 2008, 22, 2786–2798. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Xu, R.M.; Zhang, Y.; Struhl, K. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J. Biol. Chem. 2002, 277, 34655–34657. [Google Scholar] [CrossRef]
- Sun, Z.W.; Allis, C.D. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 2002, 418, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Malovichko, Y.V.; Antonets, K.S.; Maslova, A.R.; Andreeva, E.A.; Inge-Vechtomov, S.G.; Nizhnikov, A.A. RNA Sequencing Reveals Specific TranscriptomicSignatures Distinguishing Effects of the [SWI(+)] Prion and SWI1 Deletion in Yeast Saccharomyces cerevisiae. Genes 2019, 10, 212. [Google Scholar] [CrossRef]
- Treusch, S.; Lindquist, S. An intrinsically disordered yeast prion arrests the cell cycle by sequestering a spindle pole body component. J. Cell Biol. 2012, 197, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041–1072. [Google Scholar] [CrossRef]
- Du, Z.; Goncharoff, D.K.; Cheng, X.; Li, L. Analysis of [SWI(+) ] formation and propagation events. Mol. Microbiol. 2017, 104, 105–124. [Google Scholar] [CrossRef]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef]
- Bennett, S.A.; Cobos, S.N.; Meykler, M.; Fallah, M.; Rana, N.; Chen, K.; Torrente, M.P. Characterizing Histone Post-translational Modification Alterations in Yeast Neurodegenerative Proteinopathy Models. J. Vis. Exp. 2019, 145, e59104. [Google Scholar] [CrossRef]
- Alberti, S.; Halfmann, R.; Lindquist, S. Biochemical, cell biological, and genetic assays to analyze amyloid and prion aggregation in yeast. Methods Enzymol. 2010, 470, 709–734. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, O.; Suda, H.; Ohtani, T.; Sone, H. Molecular cloning and analysis of the dominant flocculation gene FLO8 from Saccharomyces cerevisiae. Mol. Gen. Genet. 1996, 251, 707–715. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [CrossRef]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobos, S.N.; Janani, C.; Cruz, G.; Rana, N.; Son, E.; Frederic, R.; Paredes Casado, J.; Khan, M.; Bennett, S.A.; Torrente, M.P. [PRION+] States Are Associated with Specific Histone H3 Post-Translational Modification Changes. Pathogens 2022, 11, 1436. https://doi.org/10.3390/pathogens11121436
Cobos SN, Janani C, Cruz G, Rana N, Son E, Frederic R, Paredes Casado J, Khan M, Bennett SA, Torrente MP. [PRION+] States Are Associated with Specific Histone H3 Post-Translational Modification Changes. Pathogens. 2022; 11(12):1436. https://doi.org/10.3390/pathogens11121436
Chicago/Turabian StyleCobos, Samantha N., Chaim Janani, Gabriel Cruz, Navin Rana, Elizaveta Son, Rania Frederic, Jailene Paredes Casado, Maliha Khan, Seth A. Bennett, and Mariana P. Torrente. 2022. "[PRION+] States Are Associated with Specific Histone H3 Post-Translational Modification Changes" Pathogens 11, no. 12: 1436. https://doi.org/10.3390/pathogens11121436
APA StyleCobos, S. N., Janani, C., Cruz, G., Rana, N., Son, E., Frederic, R., Paredes Casado, J., Khan, M., Bennett, S. A., & Torrente, M. P. (2022). [PRION+] States Are Associated with Specific Histone H3 Post-Translational Modification Changes. Pathogens, 11(12), 1436. https://doi.org/10.3390/pathogens11121436