
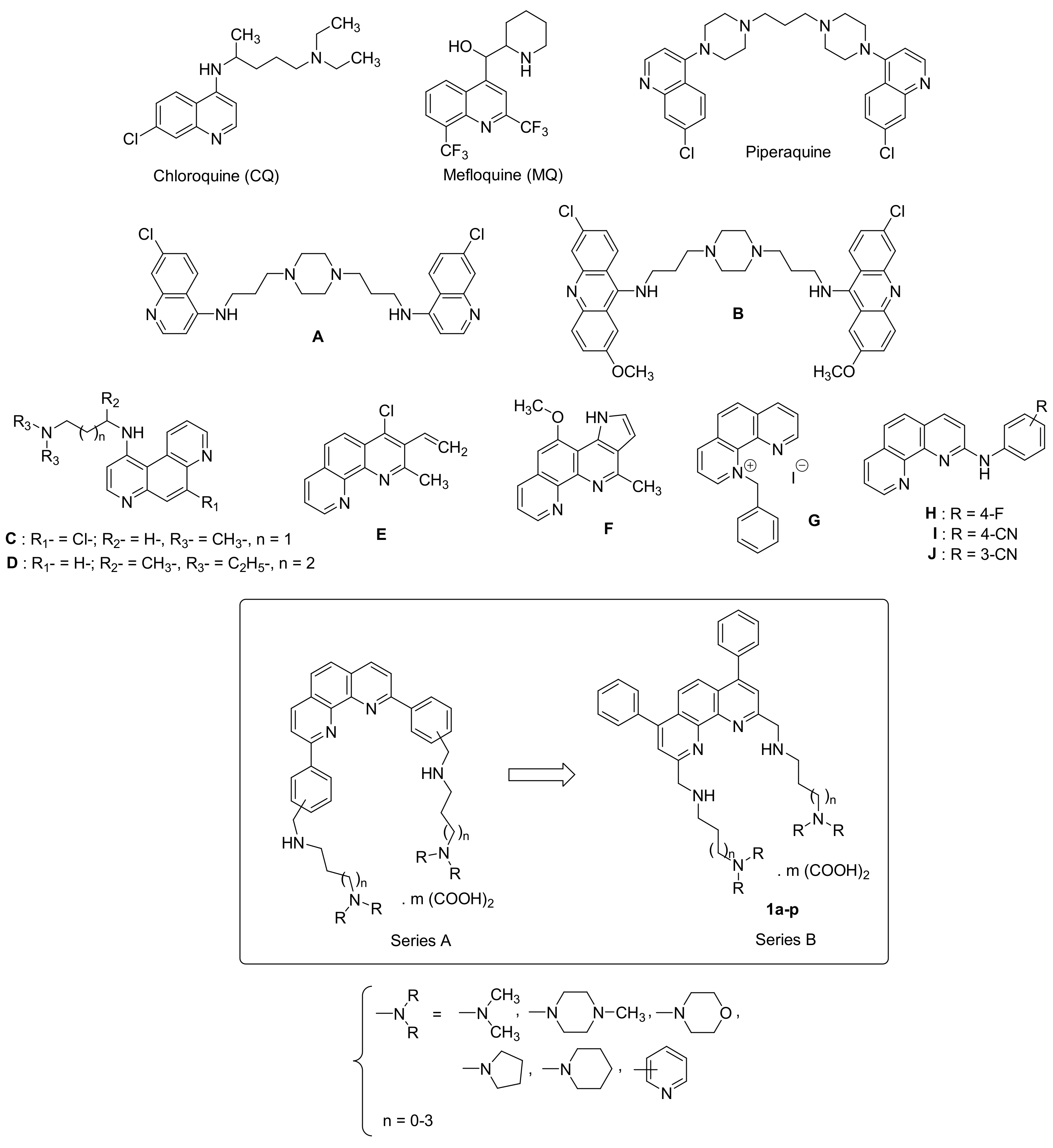
Design, Synthesis, and Antiprotozoal Evaluation of New Promising 2,9-Bis[(substituted-aminomethyl)]-4,7-phenyl-1,10-phenanthroline Derivatives, a Potential Alternative Scaffold to Drug Efflux

 , , , , , , add
Show full author list
, , , , , , add
Show full author list
Abstract
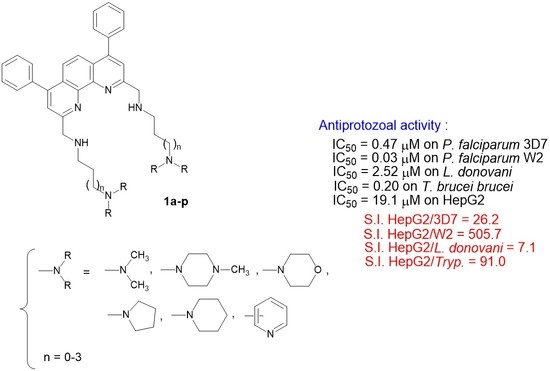
:
1. Introduction
2. Materials and Methods
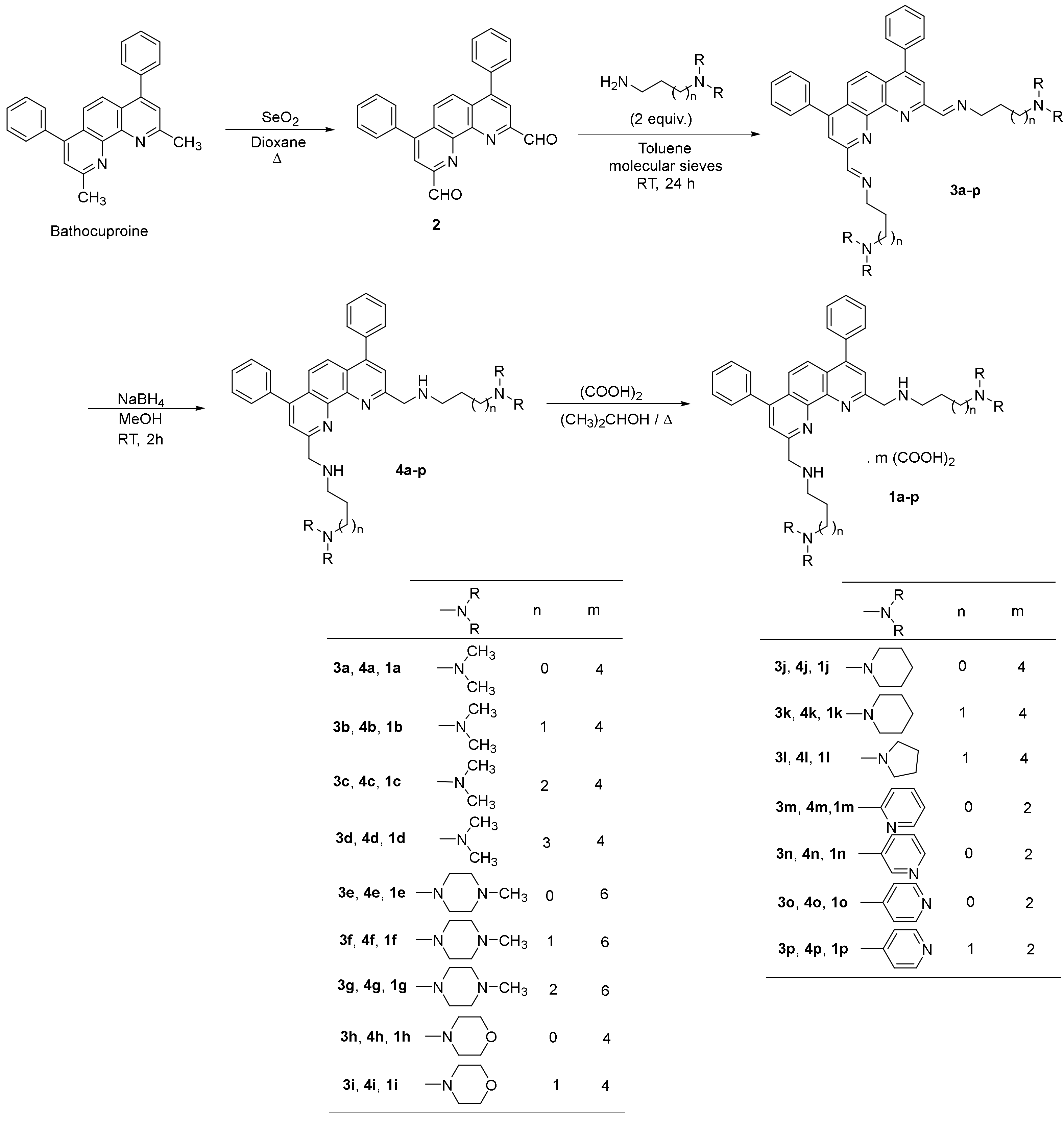
2.1. Chemistry
2.1.1. General
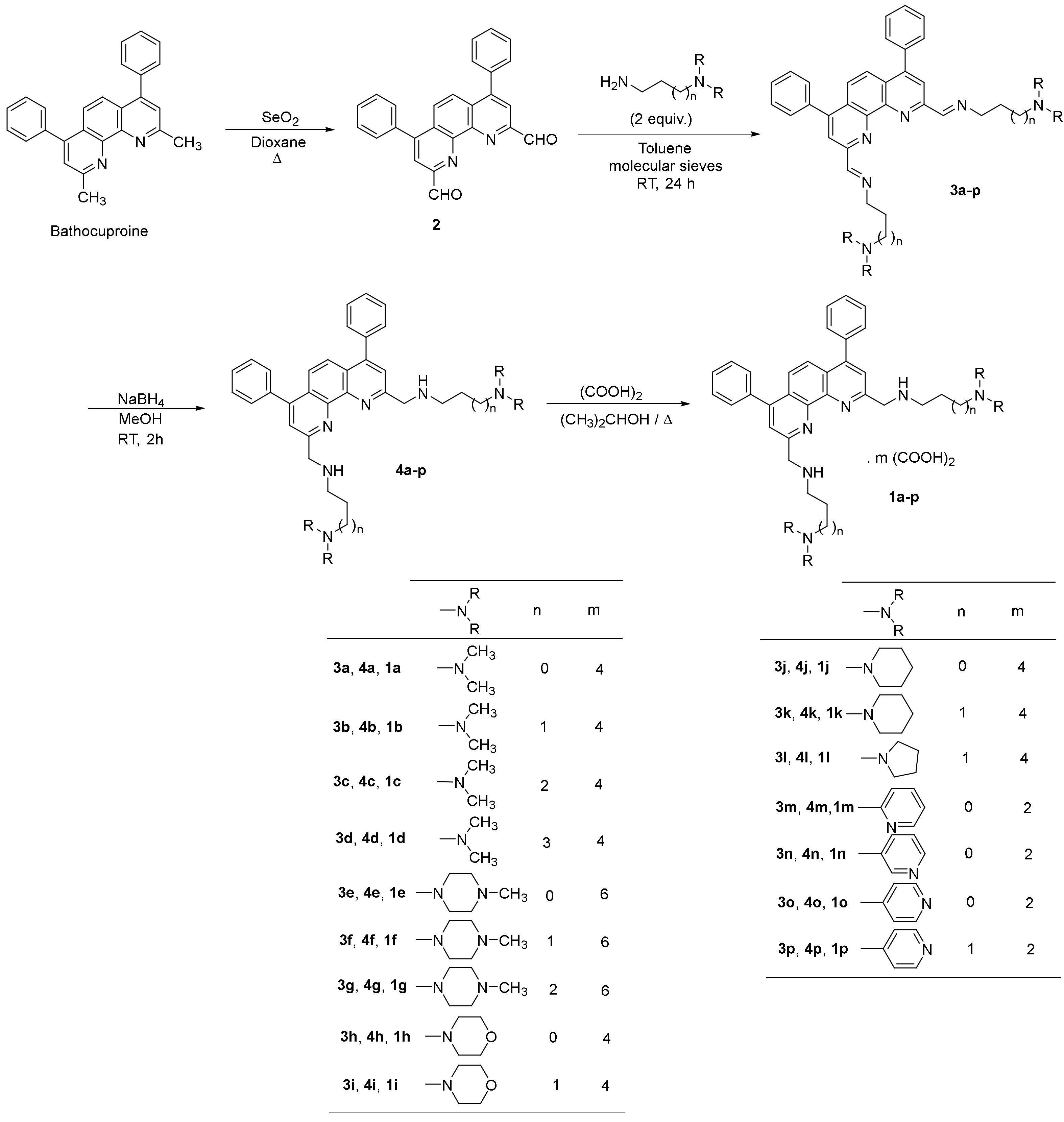
2.1.2. Synthesis of the 2,9-bis(formyl)-4,7-diphenyl-1,10-phenanthroline (2)
2.1.3. General Procedure for the Synthesis of 2,9-Bis[(substituted-iminomethyl)phenyl]-4,7-diphenyl-1,10-phenanthrolines (3a-p)
2,9-Bis[(2-dimethylaminoethyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3a)
2,9-Bis[(3-dimethylaminopropyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3b)
2,9-Bis[(4-dimethylaminobutyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3c)
2,9-Bis[(5-dimethylaminopentyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3d)
2,9-Bis [2-(4-methylpiperazin-1-yl)ethyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3e)
2,9-Bis[[(3-(4-methylpiperazin-1-yl)propyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3f)
2,9-Bis[[(4-(4-methylpiperazin-1-yl)butyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3g)
2,9-Bis [2-(morpholin-1-yl)ethyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3h)
2,9-Bis [3-(morpholin-1-yl)propyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3i)
2,9-Bis [2-(piperidin-1-yl)ethyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3j)
2,9-Bis [3-(piperidin-1-yl)propyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3k)
2,9-Bis [3-(pyrrolidin-1-yl)propyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3l)
2,9-Bis [2-(pyridin-2-yl)ethyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3m)
2,9-Bis [2-(pyridin-3-yl)ethyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3n)
2,9-Bis [2-(pyridin-4-yl)ethyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3o)
2,9-Bis [3-(pyridin-4-yl)propyl)iminomethyl]-4,7-diphenyl-1,10-phenanthroline (3p)
2.1.4. General Procedure for the Synthesis of 2,9-Bis[(substituted-aminomethyl)phenyl]-4,7-diphenyl-1,10-phenanthrolines 4a-p
2,9-Bis[(2-dimethylaminoethyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4a)
2,9-Bis[(3-dimethylaminopropyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4b)
2,9-Bis[(4-dimethylaminobutyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4c)
2,9-Bis[(5-dimethylaminopentyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4d)
2,9-Bis [2-(4-methylpiperazin-1-yl)ethyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4e)
2,9-Bis[[(3-(4-methylpiperazin-1-yl)propyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4f)
2,9-Bis[[(4-(4-methylpiperazin-1-yl)butyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4g)
2,9-Bis [2-(morpholin-1-yl)ethyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4h)
2,9-Bis [3-(morpholin-1-yl)propyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4i)
2,9-Bis [2-(piperidin-1-yl)ethyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4j)
2,9-Bis [3-(piperidin-1-yl)propyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4k)
2,9-Bis [3-(pyrrolidin-1-yl)propyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4l)
2,9-Bis [2-(pyridin-2-yl)ethyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4m)
2,9-Bis [2-(pyridin-3-yl)ethyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4n)
2,9-Bis [2-(pyridin-4-yl)ethyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4o)
2,9-Bis [3-(pyridin-4-yl)propyl)aminomethyl]-4,7-diphenyl-1,10-phenanthroline (4p)
2.1.5. General procedure for 2,9-bis[(substituted-aminomethyl)phenyl]-4,7-diphenyl-1,10-phenanthrolines 1a-p
2.2. Biological Evaluation
2.2.1. In Vitro Antiplasmodial Activity
2.2.2. In Vitro Antileishmanial Activity
2.2.3. In Vitro Antitrypanosomal Activity
2.2.4. Cytotoxicity Evaluation
2.3. FRET Melting Experiments
2.4. Native Electrospray Mass Spectrometry and Circular Dichroism
2.4.1. Samples
2.4.2. Native Mass Electrospray Spectrometry
2.4.3. Circular Dichroism
3. Results
3.1. Chemistry
3.2. Biological Evaluation
3.2.1. In Vitro Antimalarial Activity
3.2.2. In Vitro Antileishmanial Activity against Promastigote Forms
3.2.3. In Vitro Activity against Trypanosoma Brucei Brucei
3.2.4. Cytotoxicity and Selectivity Index
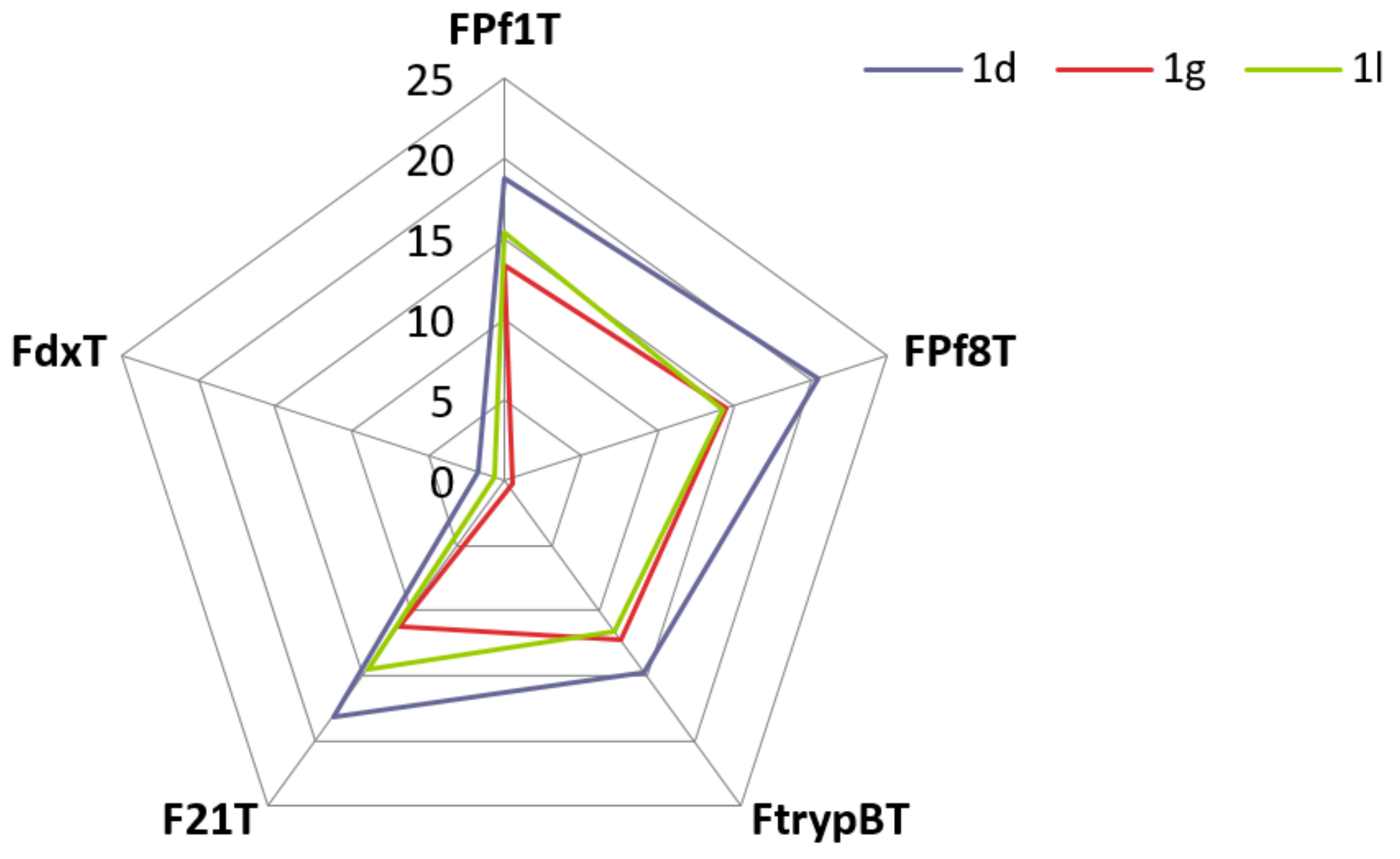
3.3. FRET Melting Experiments
3.4. Native Electrospray Mass Spectrometry and Circular Dichroism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Malaria Programme: Tools for Monitoring Antimalarial Drug Efficacy. Available online: https://www.who.int/teams/global-malaria-programme/case-management/drug-efficacy-and-resistance/tools-for-monitoring-antimalarial-drug-efficacy (accessed on 16 August 2022).
- World malaria report 2021. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021 (accessed on 16 August 2022).
- WHO Guidelines for Malaria. Available online: https://apps.who.int/iris/handle/10665/354781 (accessed on 16 August 2022).
- WHO Recommends Groundbreaking Malaria Vaccine for Children at Risk. Available online: https://www.who.int/news/item/06-10-2021-who-recommends-groundbreaking-malaria-vaccine-for-children-at-risk (accessed on 16 August 2022).
- Malaria Chemoprevention Efficacy Study Protocol. Available online: https://www.who.int/publications/i/item/9789240054769 (accessed on 16 August 2022).
- Belete, T.M. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des. Devel. Ther. 2020, 14, 3875–3889. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.K. 8-Aminoquinoline Therapy for Latent Malaria. Clin. Microbiol. Rev. 2019, 32, e00011-19. [Google Scholar] [CrossRef] [PubMed]
- Dola, V.R.; Soni, A.; Agarwal, P.; Ahmad, H.; Rama Raju, K.S.; Rashid, M.; Wahajuddin, M.; Srivastava, K.; Haq, W.; Dwivedi, A.K.; et al. Synthesis and Evaluation of Chirally Defined Side Chain Variants of 7-Chloro-4-Aminoquinoline to Overcome Drug Resistance in Malaria Chemotherapy. Antimicrob. Agents Chemother. 2017, 61, e01152-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manohar, S.; Tripathi, M.; Rawat, D.S. 4-Aminoquinoline based molecular hybrids as antimalarials: An overview. Curr. Top. Med. Chem. 2014, 14, 1706–1733. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.M.; Ward, S.A.; Berry, N.G.; Jeyadevan, J.P.; Biagini, G.A.; Asadollaly, E.; Park, B.K.; Bray, P.G. A medicinal chemistry perspective on 4-aminoquinoline antimalarial drugs. Curr. Top. Med. Chem. 2006, 6, 479–507. [Google Scholar]
- Krogstad, D.J.; Gluzman, I.Y.; Kyle, D.E.; Oduola, A.M.; Martin, S.K.; Milhous, W.K.; Schlesinger, P.H. Efflux of chloroquine from Plasmodium falciparum: Mechanism of chloroquine resistance. Science 1987, 238, 1283–1285. [Google Scholar] [CrossRef]
- Fidock, D.A.; Nomura, T.; Talley, A.K.; Cooper, R.A.; Dzekunov, S.M.; Ferdig, M.T.; Ursos, L.M.; Sidhu, A.B.; Naudé, B.; Deitsch, K.W.; et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell. 2000, 6, 861–871. [Google Scholar] [CrossRef]
- Roepe, P.D. Molecular and physiologic basis of quinoline drug resistance in Plasmodium falciparum malaria. Future Microbiol. 2009, 4, 441–455. [Google Scholar] [CrossRef]
- Deshpande, S.; Kuppast, B. 4-aminoquinolines: An Overview of Antimalarial Chemotherapy. Med. Chem. 2016, 6, 1. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, R.K.; Patial, B.; Goyal, S.; Bhardwaj, T.R. Recent advances in novel heterocyclic scaffolds for the treatment of drug-resistant malaria. J. Enzyme Inhib. Med. Chem. 2016, 31, 173–186. [Google Scholar] [CrossRef]
- Van de Walle, T.; Cools, L.; Mangelinckx, S.; D’hooghe, M. Recent contributions of quinolines to antimalarial and anticancer drug discovery research. Eur. J. Med. Chem. 2021, 226, 113865. [Google Scholar] [CrossRef]
- Douglas, B.; Kermack, W.O. Attempts to find new antimalarials. Part XXVIII. p-Phenanthroline derivatives substituted in the 4-position. J. Chem. Soc. 1949, 1017–1022. [Google Scholar] [CrossRef]
- Yapi, A.-D.; Valentin, A.; Chezal, J.-M.; Chavignon, O.; Chaillot, B.; Gerhardt, R.; Teulade, J.-C.; Blache, Y. In Vitro and in Vivo Antimalarial Activity of Derivatives of 1,10-Phenanthroline Framework. Arch. Pharm. Chem. Life Sci. 2006, 339, 201–206. [Google Scholar] [CrossRef]
- Sall, C.; Yapi, A.-D.; Desbois, A.N.; Chevalley, S.; Chezal, J.-M.; Tan, K.; Teulade, J.-C.; Valentin, A.; Blache, Y. Design, synthesis, and biological activities of conformationally restricted analogs of primaquine with a 1,10-phenanthroline framework. Bioorg. Med. Chem. Lett. 2008, 18, 4666–4669. [Google Scholar] [CrossRef]
- Wijayanti, M.A.; Sholikhah, E.N.; Hadanu, R.; Jumina, J.; Supargiyono, S.; Mustofa, M. Additive in vitro antiplasmodial effect of N-alkyl and N-benzyl-1,10-phenanthroline derivatives and cysteine protease inhibitor e64. Malar. Res. Treat. 2010, 2010, 540786. [Google Scholar] [CrossRef] [Green Version]
- Sholikhah, E.N.; Supargiyono, S.; Jumina, J.; Wijayanti, M.A.; .Tahir, I.; Hadanu, R.; Mustofa, M. In vitro antiplasmodial activity and cytotoxicity of newly synthesized N-alkyl and N-benzyl-1,10-phenanthroline derivatives. Southeast Asian J. Trop. Med. Public Health 2006, 37, 1072–1077. [Google Scholar]
- Zuma, A.A.; da Silva, R.B.; Garden, S.J.; de Souza, W. In vitro study of the trypanocidal activity of anilinophenanthrolines against Trypanosoma cruzi. Parasitol. Int. 2021, 83, 102338. [Google Scholar] [CrossRef]
- Ending the Neglect to Attain the Sustainable Development Goals: A Road Map for Neglected Tropical Diseases 2021–2030: : Overview. Available online: https://www.who.int/publications/i/item/9789240010352 (accessed on 16 August 2022).
- Fernandez-Prada, C.; Minguez-Menendez, A.; . Pena, J.; Tunes, L.G.; Pires, D.E.V.; Monte-Neto, R. Repurposed molecules: A new hope in tackling neglected infectious diseases. In Silico Drug Design: Repurposing Techniques and Methodologies, 1st ed.; Roy, K., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 119–160. [Google Scholar]
- Guillon, J.; Grellier, P.; Labaied, M.; Sonnet, P.; Léger, J.-M.; Déprez-Poulain, R.; Forfar-Bares, I.; Dallemagne, P.; Lemaître, N.; Péhourcq, F.; et al. Synthesis, antimalarial activity, and molecular modeling of new pyrrolo [1,2-a]quinoxalines, bispyrrolo [1,2-a]quinoxalines, bispyrido [3,2-e]pyrrolo [1,2-a]pyrazines, and bispyrrolo [1,2-a]thieno [3,2-e]pyrazines. J. Med. Chem. 2004, 47, 1997–2009. [Google Scholar] [CrossRef]
- Dassonville-Klimpt, A.; Cézard, C.; Mullié, C.; Agnamey, P.; Jonet, A.; Da Nascimento, S.; Marchivie, M.; Guillon, J.; Sonnet, P. Absolute Configuration and Antimalarial Activity of erythro-Mefloquine Enantiomers. ChemPlusChem 2013, 78, 642–646. [Google Scholar] [CrossRef]
- Guillon, J.; Cohen, A.; Gueddouda, N.M.; Das, R.N.; Moreau, S.; Ronga, L.; Savrimoutou, S.; Basmaciyan, L.; Monnier, A.; Monget, M.; et al. Design, synthesis and antimalarial activity of novel bis{N-[(pyrrolo [1,2-a]quinoxalin-4-yl)benzyl]-3-aminopropyl}amine derivatives. J. Enzyme Inhib. Med. Chem. 2017, 32, 547–563. [Google Scholar] [CrossRef] [Green Version]
- Jonet, A.; Guillon, J.; Mullie, C.; Cohen, A.; Bentzinger, G.; Schneider, J.; Taudon, N.; Hutter, S.; Azas, N.; Moreau, S.; et al. Synthesis and Antimalarial Activity of New Enantiopure Aminoalcoholpyrrolo [1,2-a]quinoxalines. Med. Chem. 2018, 14, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Guillon, J.; Cohen, A.; Boudot, C.; Valle, A.; Milano, V.; Das, R.N.; Guédin, A.; Moreau, S.; Ronga, L.; Savrimoutou, S.; et al. Design, synthesis, and antiprotozoal evaluation of new 2,4-bis[(substituted-aminomethyl)phenyl]quinoline, 1,3-bis[(substituted-aminomethyl)phenyl]isoquinoline and 2,4-bis[(substituted-aminomethyl)phenyl]quinazoline derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 432–459. [Google Scholar] [CrossRef] [PubMed]
- Dassonville-Klimpt, A.; Schneider, J.; Damiani, C.; Tisnerat, C.; Cohen, A.; Azas, N.; Marchivie, M.; Guillon, J.; Mullié, C.; Agnamey, P.; et al. Design, synthesis, and characterization of novel aminoalcohol quinolines with strong in vitro antimalarial activity. Eur. J. Med. Chem. 2022, 228, 113981. [Google Scholar] [CrossRef] [PubMed]
- Guillon, J.; Cohen, A.; Nath Das, R.; Boudot, C.; Meriem Gueddouda, N.; Moreau, S.; Ronga, L.; Savrimoutou, S.; Basmaciyan, L.; Tisnerat, C.; et al. Design, synthesis, and antiprotozoal evaluation of new 2,9-bis[(substituted-aminomethyl)phenyl]-1,10-phenanthroline derivatives. Chem. Biol. Drug Des. 2018, 91, 974–995. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.P.; Wasserman, M. G-Quadruplex ligands: Potent inhibitors of telomerase activity and cell proliferation in Plasmodium falciparum. Mol. Biochem. Parasitol. 2016, 207, 33–38. [Google Scholar] [CrossRef]
- Tidwell, R.R.; Boykin, D.W.; Ismail, M.A.; Wilson, W.D.; White, E.W.; Kumar, A.; Nanjunda, R. Dicationic Compounds Which Selectively Recognize G-Quadruplex DNA. U.S. Patent US74168905P, 20 June 2007. [Google Scholar]
- Leeder, W.-M.; Hummel, N.F.C.; Göringer, H.U. Multiple G-quartet structures in pre-edited mRNAs suggest evolutionary driving force for RNA editing in trypanosomes. Sci. Rep. 2016, 6, 29810. [Google Scholar] [CrossRef] [Green Version]
- Lombrana, R.; Alvarez, A.; Fernandez-Justel, J.M.; Almeida, R.; Poza-Carrion, C.; Gomes, F.; Calzada, A.; Requena, J.M.; Gomez, M. Transcriptionally Driven DNA Replication Program of the Human Parasite Leishmania major. Cell Rep. 2016, 16, 1774–1786. [Google Scholar] [CrossRef] [Green Version]
- Bottius, E.; Bakhsis, N.; Scherf, A. Plasmodium falciparum Telomerase: De Novo Telomere Addition to Telomeric and Nontelomeric Sequences and Role in Chromosome Healing. Mol. Cell. Biol. 1998, 18, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Raj, D.K.; Das, D.R.; Dash, A.P.; Supakar, P.C. Identification of telomerase activity in gametocytes of Plasmodium falciparum. Biochem. Biophys. Res. Commun. 2003, 309, 685–688. [Google Scholar] [CrossRef]
- De Cian, A.; Grellier, P.; Mouray, E.; Depoix, D.; Bertrand, H.; Monchaud, D.; Telade-Fichou, M.-P.; Mergny, J.-L.; Alberti, P. Plasmodium Telomeric Sequences: Structure, Stability and Quadruplex Targeting by Small Compounds. ChemBioChem 2008, 9, 2730–2739. [Google Scholar] [CrossRef]
- Desjardins, R.E.; Canfield, C.J.; Haynes, J.D.; Chulay, J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Bennett, T.N.; Paguio, M.; Gligorijevic, B.; Seudieu, C.; Kosar, A.D.; Davidson, E.; Roepe, P.D. Novel, Rapid, and Inexpensive Cell-Based Quantification of Antimalarial Drug Efficacy. Antimicrob. Agents Chemother. 2004, 48, 1807–1810. [Google Scholar] [CrossRef]
- Bacon, D.J.; Latour, C.; Lucas, C.; Colina, O.; Ringwald, P.; Picot, S. Comparison of a SYBR Green I-Based Assay with a Histidine-Rich Protein II Enzyme-Linked Immunosorbent Assay for In Vitro Antimalarial Drug Efficacy Testing and Application to Clinical Isolates. Antimicrob. Agents Chemother. 2007, 51, 1172–1178. [Google Scholar] [CrossRef] [Green Version]
- Kaddouri, H.; Nakache, S.; Houzé, S.; Mentré, F.; Le Bras, J. Assessment of the Drug Susceptibility of Plasmodium falciparum Clinical Isolates from Africa by Using a Plasmodium Lactate Dehydrogenase Immunodetection Assay and an Inhibitory Maximum Effect Model for Precise Measurement of the 50-Percent Inhibitory Concentration. Antimicrob. Agents Chemother. 2006, 50, 3343–3349. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Emami, S.A.; Zamanai Taghizadeh Rabe, S.; Ahi, A.; Mahmoudi, M. Inhibitory Activity of Eleven Artemisia Species from Iran against Leishmania Major Parasites. Iran J. Basic Med. Sci. 2012, 15, 807–811. [Google Scholar]
- Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [Google Scholar] [CrossRef]
- Baltz, T.; Baltz, D.; Giroud, C. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J. 1985, 4, 1273–1277. [Google Scholar] [CrossRef]
- De Cian, A.; Guittat, L.; Kaiser, M.; Saccà, B.; Amrane, S.; Bourdoncle, A.; Alberti, P.; Teulade-Fichou, M.-P.; Lacroix, L.; Mergny, J.-L. Fluorescence-based melting assays for studying quadruplex ligands. Methods 2007, 42, 183–195. [Google Scholar] [CrossRef]
- Gabelica, V.; Livet, S.; Rosu, F. Optimizing Native Ion Mobility Q-TOF in Helium and Nitrogen for Very Fragile Noncovalent Structures. J. Am. Soc. Mass Spectrom. 2018, 29, 2189–2198. [Google Scholar] [CrossRef] [Green Version]
- Guillon, J.; Denevault-Sabourin, C.; Chevret, E.; Brachet-Botineau, M.; Milano, V.; Guédin-Beaurepaire, A.; Moreau, S.; Ronga, L.; Savrimoutou, S.; Rubio, S.; et al. Design, synthesis, and antiproliferative effect of 2,9-bis [4-(pyridinylalkylaminomethyl)phenyl]-1,10-phenanthroline derivatives on human leukemic cells by targeting G-quadruplex. Arch. Pharm. (Weinh.) 2021, 354, e2000450. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Gabelica, V. Native Hydrogen/Deuterium Exchange Mass Spectrometry of Structured DNA Oligonucleotides. Anal. Chem. 2020, 92, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.-Y.; Xia, L.-M.; Lennox, A.J.J.; Sun, J.J.; Chen, H.; Jin, H.-M.; Junge, H.; Wu, Q.-A.; Jia, J.-H.; Beller, M.; et al. Structure-Activated Copper Photosensitisers for Photocatalytic Water Reduction. Chem. Eur. J. 2017, 23, 3631–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch. Toxicol. 2018, 92, 893–906. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Donato, M.T.; Boobis, A.; Edwards, R.J.; Watts, P.S.; Castell, J.V.; Gómez-Lechón, M.J. Cytochrome P450 expression in human hepatocytes and hepatoma cell lines: Molecular mechanisms that determine lower expression in cultured cells. Xenobiotica 2002, 32, 505–520. [Google Scholar] [CrossRef]
- Largy, E.; König, A.; Ghosh, A.; Ghosh, D.; Benabou, S.; Rosu, F.; Gabelica, V. Mass Spectrometry of Nucleic Acid Noncovalent Complexes. Chem. Rev. 2022, 122, 7720–7839. [Google Scholar] [CrossRef]
- Ghosh, A.; Largy, E.; Gabelica, V. DNA G-quadruplexes for native mass spectrometry in potassium: A database of validated structures in electrospray-compatible conditions. Nucleic Acids Res. 2021, 49, 2333–2345. [Google Scholar] [CrossRef]
- Largy, E.; Marchand, A.; Amrane, S.; Gabelica, V.; Mergny, J.-L. Quadruplex Turncoats: Cation-Dependent Folding and Stability of Quadruplex-DNA Double Switches. J. Am. Chem. Soc. 2016, 138, 2780–2792. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Trajkovski, M.; Teulade-Fichou, M.-P.; Gabelica, V.; Plavec, J. Phen-DC3 Induces Refolding of Human Telomeric DNA into a Chair-Type Antiparallel G-Quadruplex through Ligand Intercalation. Angew. Chem. Int. Ed. 2022, 61, e202207384. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Salt a | mp (°C) b | % Yield c | |
|---|---|---|---|---|
| 1a | Orange crystals | 4 (COOH)2 | 179–181 | 64 |
| 1b | Beige crystals | 4 (COOH)2 | 161–163 | 60 |
| 1c | Orange crystals | 4 (COOH)2 | 111–113 | 56 |
| 1d | Beige crystals | 4 (COOH)2 | 119–121 | 57 |
| 1e | Orange crystals | 6 (COOH)2 | 185–187 | 81 |
| 1f | Yellow-orange crystals | 6 (COOH)2 | 205–207 | 58 |
| 1g | Beige crystals | 6 (COOH)2 | 207–209 | 63 |
| 1h | Beige crystals | 4 (COOH)2 | 194–196 | 73 |
| 1i | Yellow crystals | 4 (COOH)2 | 174–176 | 61 |
| 1j | Beige crystals | 4 (COOH)2 | 192–194 | 69 |
| 1k | Beige crystals | 4 (COOH)2 | 141–143 | 62 |
| 1l | Beige crystals | 4 (COOH)2 | 145–147 | 61 |
| 1m | Orange crystals | 2 (COOH)2 | 165–167 | 63 |
| 1n | Orange crystals | 2 (COOH)2 | 164–166 | 74 |
| 1o | Beige crystals | 2 (COOH)2 | 147–149 | 66 |
| 1p | Yellow crystals | 2 (COOH)2 | 99–101 | 83 |
| Compound | P. falciparum Strains IC50 Values (μM) a | L. donovani IC50 Values (μM) b | Trypanosoma brucei brucei IC50 Values (μM) c | Cytotoxicity to HepG2 Cells CC50 Values (μM) d | |
|---|---|---|---|---|---|
| W2 | 3D7 | Trypanos Antat 1.9 | |||
| CQ e | 0.40 ± 0.04 | 0.11 ± 0.01 | n.d. h | n.d. h | 30 |
| MQ e | 0.016 ± 0.002 | 0.06 ± 0.003 | n.d. h | n.d. h | n.d. h |
| Pentamidinef | n.d. h | n.d. h | 5.5 ± 0.80 | 0.0002 ± 0.00006 | 2.3 ± 0.50 |
| Amphotericin B f | n.d. h | n.d. h | 0.1 ± 0.04 | n.d. h | 8.8 ± 0.60 |
| Suramine g | n.d.h | n.d. h | n.d. h | 0.03 ± 0.003 | n.d. h |
| Fexinidazole g | n.d.h | n.d. h | n.d. h | 0.59 ± 0.039 | n.d. h |
| Eflornithineg | n.d. h | n.d. h | n.d. h | 15.19 ± 0.64 | n.d. h |
| Doxorubicin | n.d. h | n.d. h | n.d. h | n.d. h | 0.06 ± 0.02 |
| 1a | 1.00 ± 0.34 | 1.80 ± 0.67 | >12.5 i | 0.67 ± 0.03 | 6.52 ± 0.61 |
| 1b | 6.13 ± 1.63 | 21.19 ± 7.72 | >12.5 i | 0.57 ± 0.07 | 5.96 ± 0.20 |
| 1c | 0.03 ± 0.003 | 10.31 ± 2.13 | >12.5 i | 0.86 ± 0.16 | 10.43 ± 0.70 |
| 1d | 3.51 ± 0.52 | 4.79 ± 0.50 | >12.5 i | 1.42 ± 0.10 | 3.35 ± 0.33 |
| 1e | 0.92 ± 0.18 | 2.16 ± 0.32 | >12.5 i | 0.45 ± 0.03 | 10.54 ± 1.27 |
| 1f | 11.27 ± 2.93 | 21.54 ± 4.10 | >12.5 i | 0.97 ± 0.11 | 8.62 ± 0.50 |
| 1g | 2.48 ± 0.49 | 8.19 ± 3.53 | >12.5 i | 0.96 ± 0.13 | 16.75 ± 0.60 |
| 1h | 0.17 ± 0.05 | 0.67 ± 0.21 | 4.50 ± 2.20 | 0.22 ± 0.06 | 5.89 ± 0.53 |
| 1i | 0.04 ± 0.01 | 1.45 ± 0.58 | >12.5 i | 0.29 ± 0.02 | 6.29 ± 1.06 |
| 1j | 2.60 ± 0.55 | 1.59 ± 0.59 | 2.52 ± 0.20 | 0.20 ± 0.02 | 1.64 ± 0.14 |
| 1k | 1.65 ± 0.36 | 3.18 ± 0.68 | >12.5 i | 0.77 ± 0.10 | 4.29 ± 0.52 |
| 1l | 0.03 ± 0.01 | 9.91 ± 1.73 | >12.5 i | 1.13 ± 0.06 | 15.17 ± 1.56 |
| 1m | 0.07 ± 0.01 | 0.47 ± 0.12 | >12.5 i | 0.63 ± 0.01 | 4.34 ± 0.31 |
| 1n | 2.54 ± 0.36 | 0.71 ± 0.11 | 2.67 ± 0.80 | 0.43 ± 0.03 | 3.10 ± 0.19 |
| 1o | 1.87 ± 0.58 | 0.73 ± 0.19 | 2.69 ± 0.60 | 0.21 ± 0.01 | 19.11 ± 2.11 |
| 1p | 0.145 ± 0.04 | 1.02 ± 0.35 | >12.5 i | 0.65 ± 0.05 | 3.04 ± 0.19 |
| Compound | Selectivity Index a | |||
|---|---|---|---|---|
| HepG2/W2 | HepG2/3D7 | HepG2/L. donovani | HepG2/Tryp. | |
| CQ | 75 | 272 | n.d. b | n.d. b |
| Pentamidine | n.d. b | n.d. b | n.d. b | 11,500 |
| Amphotericin B | n.d. b | n.d. b | 88.0 | n.d. b |
| 1a | 6.52 | 3.62 | n.d. b | 9.73 |
| 1b | 0.97 | 0.28 | n.d. b | 10.46 |
| 1c | 347.67 | 1.01 | n.d. b | 12.13 |
| 1d | 0.95 | 0.70 | n.d. b | 2.36 |
| 1e | 11.46 | 4.88 | n.d. b | 23.42 |
| 1f | 0.76 | 0.40 | n.d. b | 8.89 |
| 1g | 6.75 | 2.04 | n.d. b | 17.45 |
| 1h | 34.64 | 8.79 | 1.31 | 26.77 |
| 1i | 157.25 | 4.34 | n.d. b | 21.69 |
| 1j | 0.63 | 1.03 | 0.65 | 8.20 |
| 1k | 2.60 | 1.35 | n.d. b | 5.57 |
| 1l | 505.7 | 1.53 | n.d. b | 13.42 |
| 1m | 62.0 | 9.23 | n.d. b | 6.89 |
| 1n | 1.22 | 4.37 | 1.16 | 7.21 |
| 1o | 10.22 | 26.18 | 7.10 | 91.0 |
| 1p | 20.97 | 2.98 | n.d. b | 4.68 |
| Compound | ΔTm (°C) a | ΔTm (°C) a | ΔTm (°C) a | ΔTm (°C) a | ΔTm (°C) a | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| FPf1T | FPf8T | FtrypBT | F21T | FdxT | ||||||
| PhenDC3 | 24.6 | ±0.1 | 24.7 | ±0.2 | 19.2 | ±0.2 | 26.3 | ±0.1 | 0.1 | ±0.2 |
| CQ | 1.9 | ±0.1 | 2.4 | ±1.2 | n.d. b | 2.4 | ±1.1 | n.d. b | ||
| MQ | 3.1 | ±0.5 | 6.6 | ±2.3 | n.d. b | 2.6 | ±0.5 | n.d. b | ||
| 1a | 1.8 | ±0.3 | 1.6 | ±0.2 | 1.9 | ±0.1 | 1.8 | ±0.4 | −0.3 | ±0.2 |
| 1b | 7.4 | ±0.6 | 7.9 | ±0.3 | 8.7 | ±0.6 | 12.8 | ±1.3 | −0.3 | ±0.2 |
| 1c | 8.9 | ±0.8 | 10.2 | ±1.0 | 10.6 | ±0.8 | 13.2 | ±0.4 | −0.5 | ±0.4 |
| 1d | 18.8 | ±0.2 | 20.5 | ±0.2 | 14.7 | ±0.7 | 18.1 | ±0.9 | 1.8 | ±0.2 |
| 1e | 9.4 | ±1.0 | 10.5 | ±0.1 | 8.0 | ±0.4 | 9.6 | ±0.6 | −1.6 | ±0.2 |
| 1f | 8.8 | ±0.5 | 10.9 | ±0.9 | 9.1 | ±0.5 | 10.8 | ±0.6 | −0.5 | ±0.1 |
| 1g | 13.4 | ±0.7 | 14.5 | ±1.2 | 12.2 | ±1.7 | 11.2 | ±0.9 | −0.5 | ±0.1 |
| 1h | 1.8 | ±0.4 | 1.9 | ±0.2 | 2.2 | ±0.1 | 2.0 | ±0.2 | −0.9 | ±0.4 |
| 1i | 6.7 | ±0.5 | 6.7 | ±0.6 | 6.0 | ±0.9 | 8.2 | ±0.1 | −1.5 | ±0.4 |
| 1j | 5.6 | ±0.6 | 7.3 | ±2.4 | 7.2 | ±0.6 | 7.7 | ±2.1 | 0.2 | ±0.1 |
| 1k | 12.8 | ±0.7 | 14.0 | ±0.3 | 8.3 | ±1.0 | 12.0 | ±0.7 | 0.4 | ±0.3 |
| 1l | 15.4 | ±0.9 | 14.2 | ±0.9 | 11.6 | ±0.9 | 14.5 | ±0.4 | 0.7 | ±0.2 |
| 1m | 1.3 | ±0.2 | 1.0 | ±0.2 | 1.8 | ±0.6 | 1.0 | ±0.5 | −0.2 | ±0.1 |
| 1n | 2.9 | ±0.4 | 5.3 | ±0.4 | 5.2 | ±0.3 | 4.6 | ±0.6 | −0.3 | ±0.1 |
| 1o | 2.9 | ±0.1 | 5.2 | ±0.1 | 5.3 | ±0.7 | 3.9 | ±0.3 | −0.3 | ±0.2 |
| 1p | 3.5 | ±0.3 | 3.9 | ±0.7 | 3.7 | ±0.3 | 3.6 | ±0.3 | −1.1 | ±0.2 |
| Ligands | Kd (µM) a | |||||
|---|---|---|---|---|---|---|
| Name | Stoichiometry | Pf1 | Pf8 | 24TTG | 24nonG4 | DK66 |
| 1c | 1 | 23 | 3 | 44 | 9 | 110 |
| 1d | 1 | 8 | 3 | 38 | 11 | 53 |
| 1l | 1 | 10 | 2 | 34 | 8 | 55 |
| 1m | 1 | n.b. | 83 | n.b. | n.b. | n.b. |
| 1c | 2 | n.b. | 50 | 28 | <LOQ | n.b. |
| 1d | 2 | 24 | 16 | 62 | <LOQ | n.b. |
| 1l | 2 | 37 | 59 | 18 | <LOQ | n.b. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillon, J.; Cohen, A.; Boudot, C.; Monic, S.; Savrimoutou, S.; Moreau, S.; Albenque-Rubio, S.; Lafon-Schmaltz, C.; Dassonville-Klimpt, A.; Mergny, J.-L.; et al. Design, Synthesis, and Antiprotozoal Evaluation of New Promising 2,9-Bis[(substituted-aminomethyl)]-4,7-phenyl-1,10-phenanthroline Derivatives, a Potential Alternative Scaffold to Drug Efflux. Pathogens 2022, 11, 1339. https://doi.org/10.3390/pathogens11111339
Guillon J, Cohen A, Boudot C, Monic S, Savrimoutou S, Moreau S, Albenque-Rubio S, Lafon-Schmaltz C, Dassonville-Klimpt A, Mergny J-L, et al. Design, Synthesis, and Antiprotozoal Evaluation of New Promising 2,9-Bis[(substituted-aminomethyl)]-4,7-phenyl-1,10-phenanthroline Derivatives, a Potential Alternative Scaffold to Drug Efflux. Pathogens. 2022; 11(11):1339. https://doi.org/10.3390/pathogens11111339
Chicago/Turabian StyleGuillon, Jean, Anita Cohen, Clotilde Boudot, Sarah Monic, Solène Savrimoutou, Stéphane Moreau, Sandra Albenque-Rubio, Camille Lafon-Schmaltz, Alexandra Dassonville-Klimpt, Jean-Louis Mergny, and et al. 2022. "Design, Synthesis, and Antiprotozoal Evaluation of New Promising 2,9-Bis[(substituted-aminomethyl)]-4,7-phenyl-1,10-phenanthroline Derivatives, a Potential Alternative Scaffold to Drug Efflux" Pathogens 11, no. 11: 1339. https://doi.org/10.3390/pathogens11111339