
Isolation and Production of Human Monoclonal Antibody Proteins against a Toxocara canis Excretory–Secretory Recombinant Antigen

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Recombinant TES-26 Protein Preparation
2.2. Recombinant TES-26 Protein Sequence Analysis
2.3. Biopanning, Phage ELISA and DNA Sequencing
2.4. Recombinant Monoclonal Antibody Protein Expression and Purification
2.5. Antigen-Antibody Binding Assays
2.5.1. Western Blot Using Recombinant and Native Antigen Proteins
2.5.2. Immunoassays Using Recombinant and Native Antigen Proteins
2.6. Titration ELISA
2.7. Specificity ELISA
2.8. Surface Plasmon Resonance Analysis
2.9. Preliminary Antigen Detection ELISA Using Human Serum Samples
3. Results
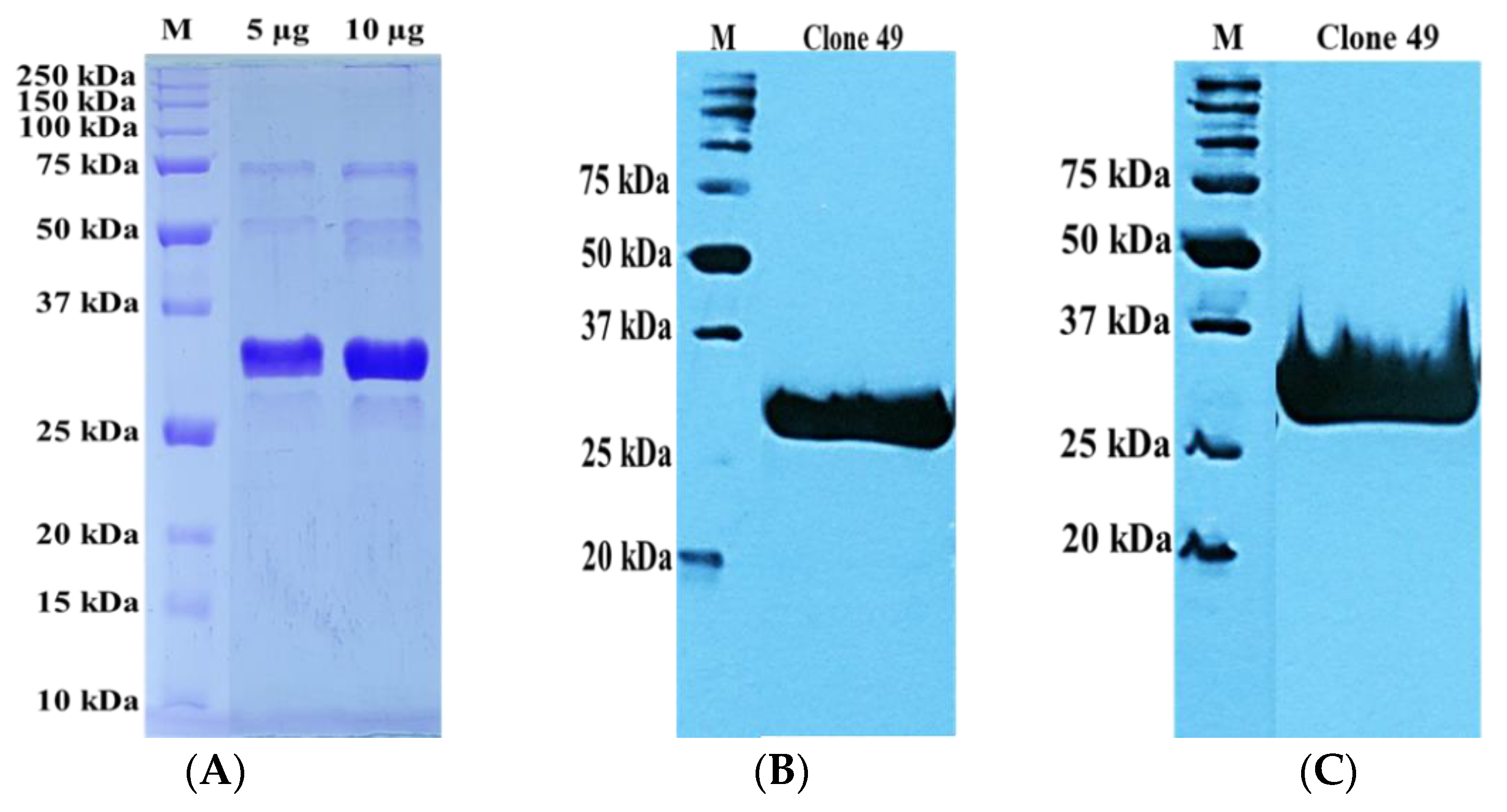
3.1. Recombinant TES-26 Protein Preparation, Verification and Sequence Analysis
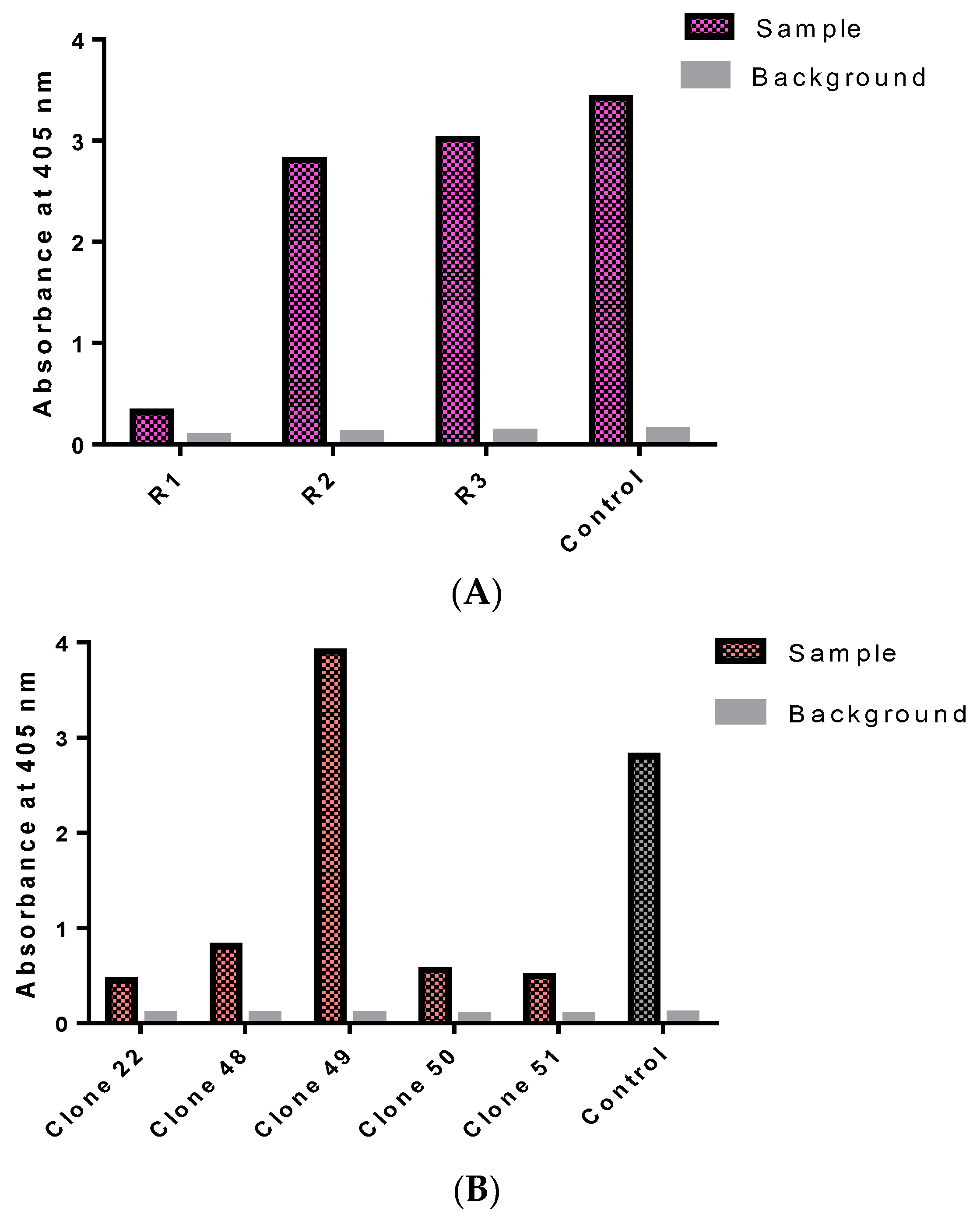
3.2. Isolation of Monoclonal Antibodies
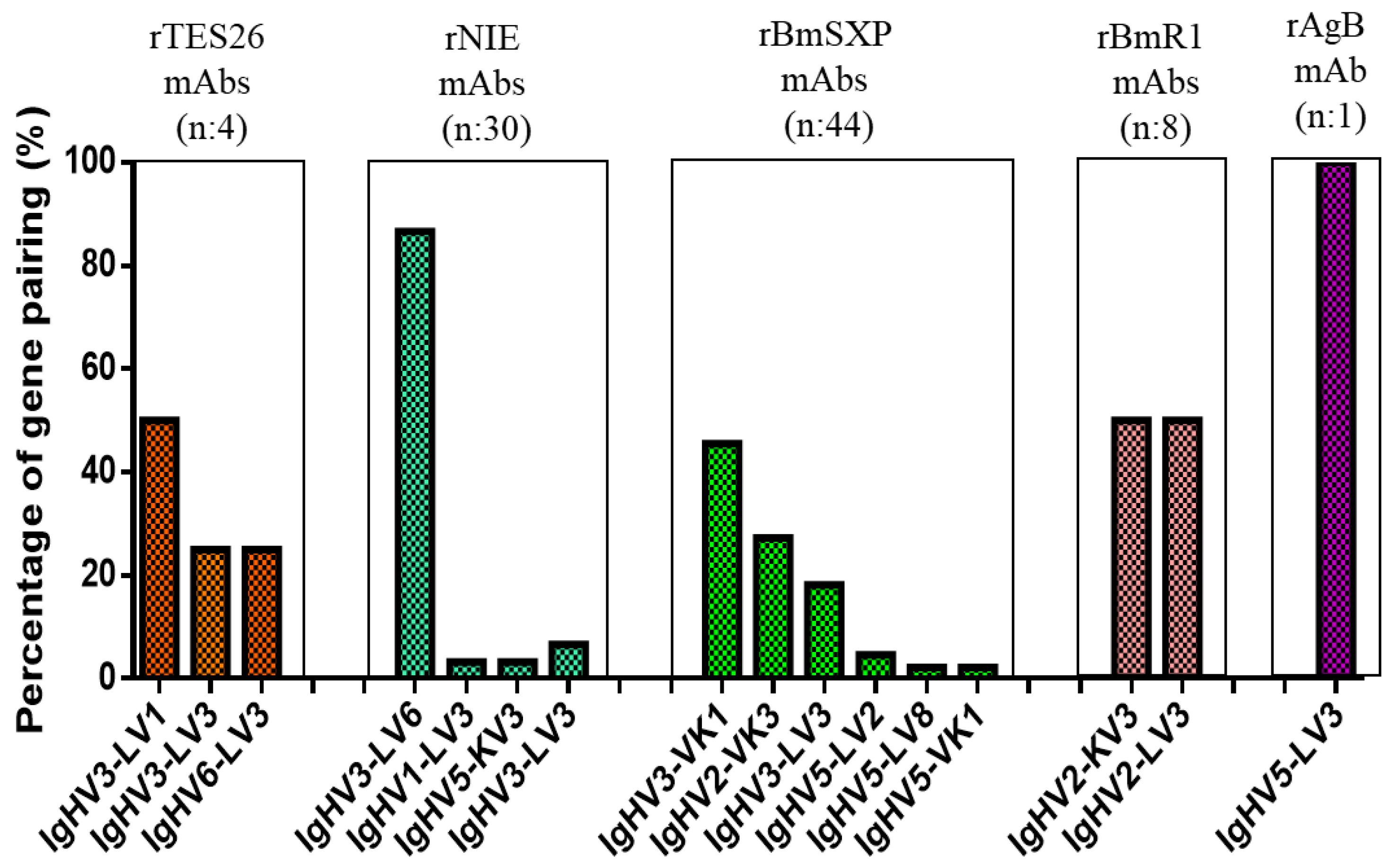
3.3. Monoclonal Antibody Gene Analysis
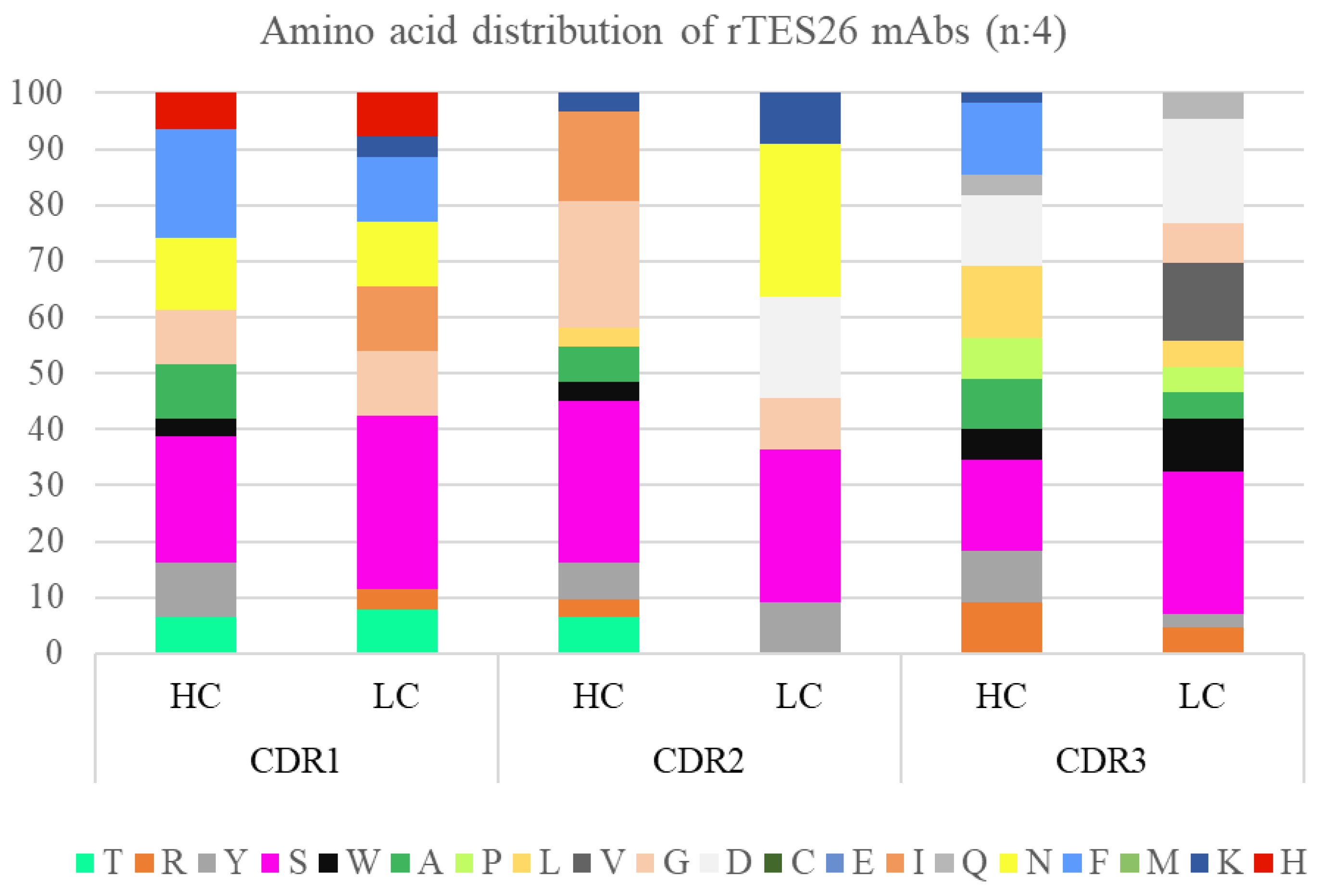
3.4. Monoclonal Antibody Gene Sequence Analysis
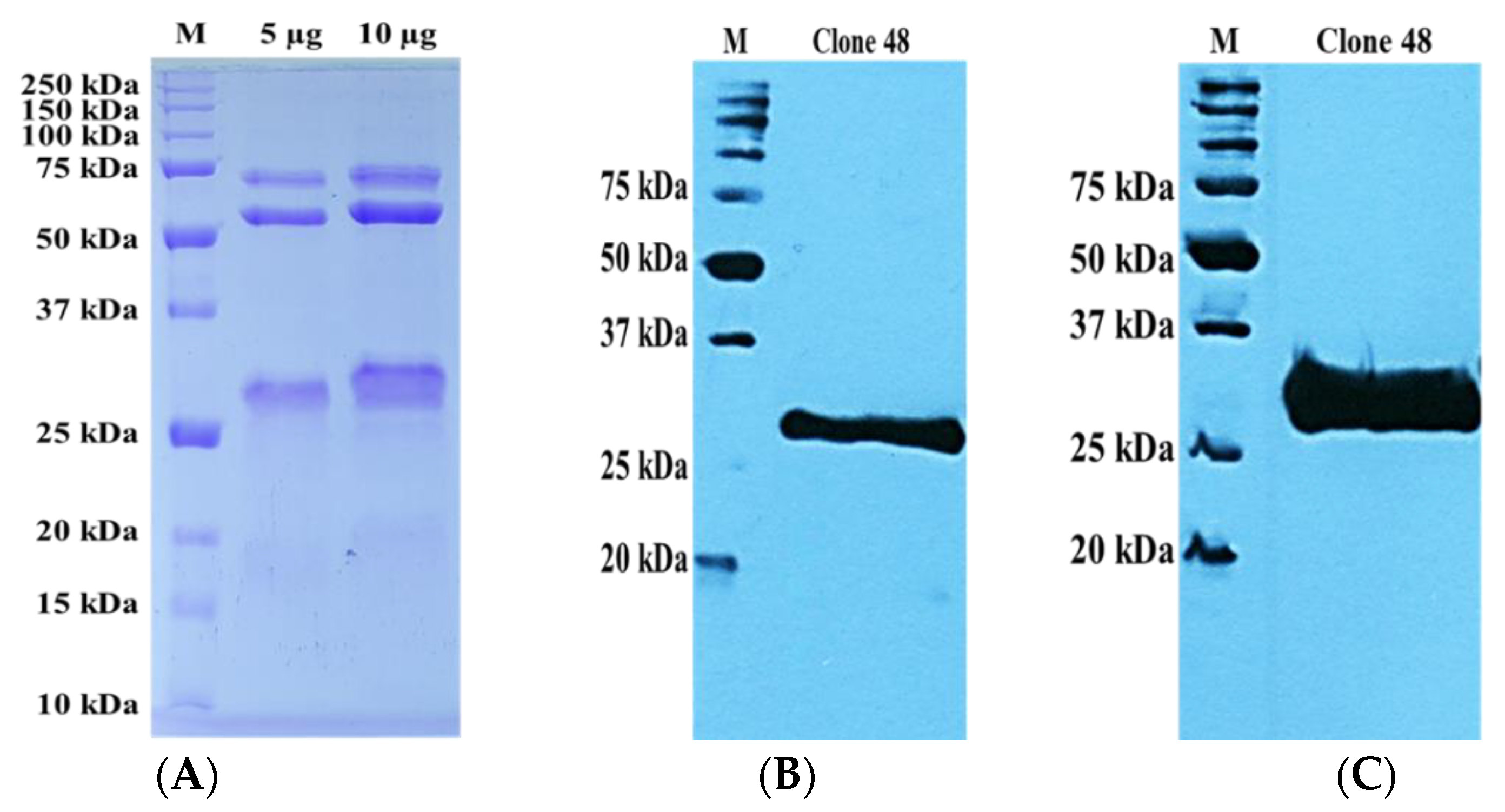
3.5. Preparation of Recombinant Monoclonal Antibody Protein
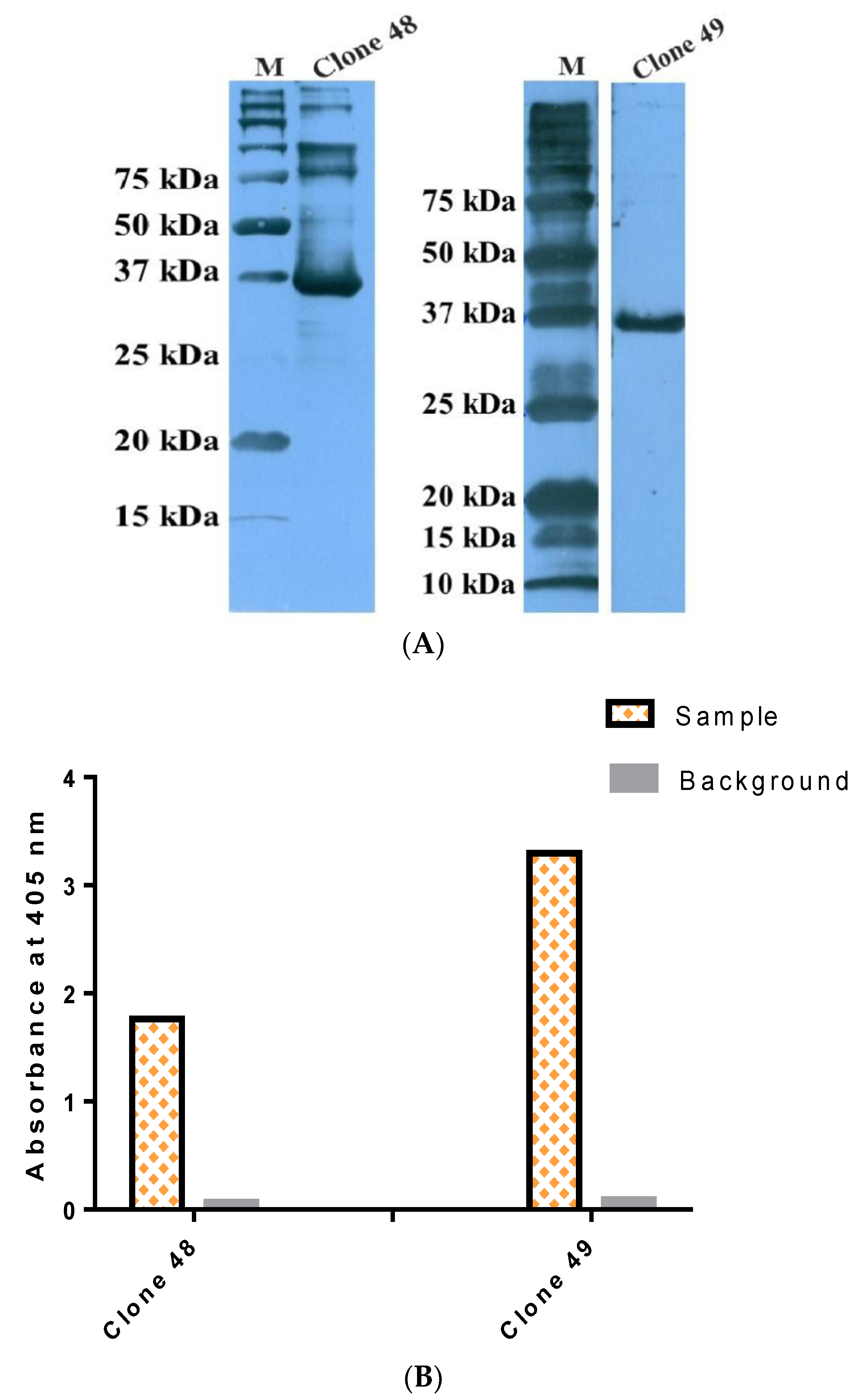
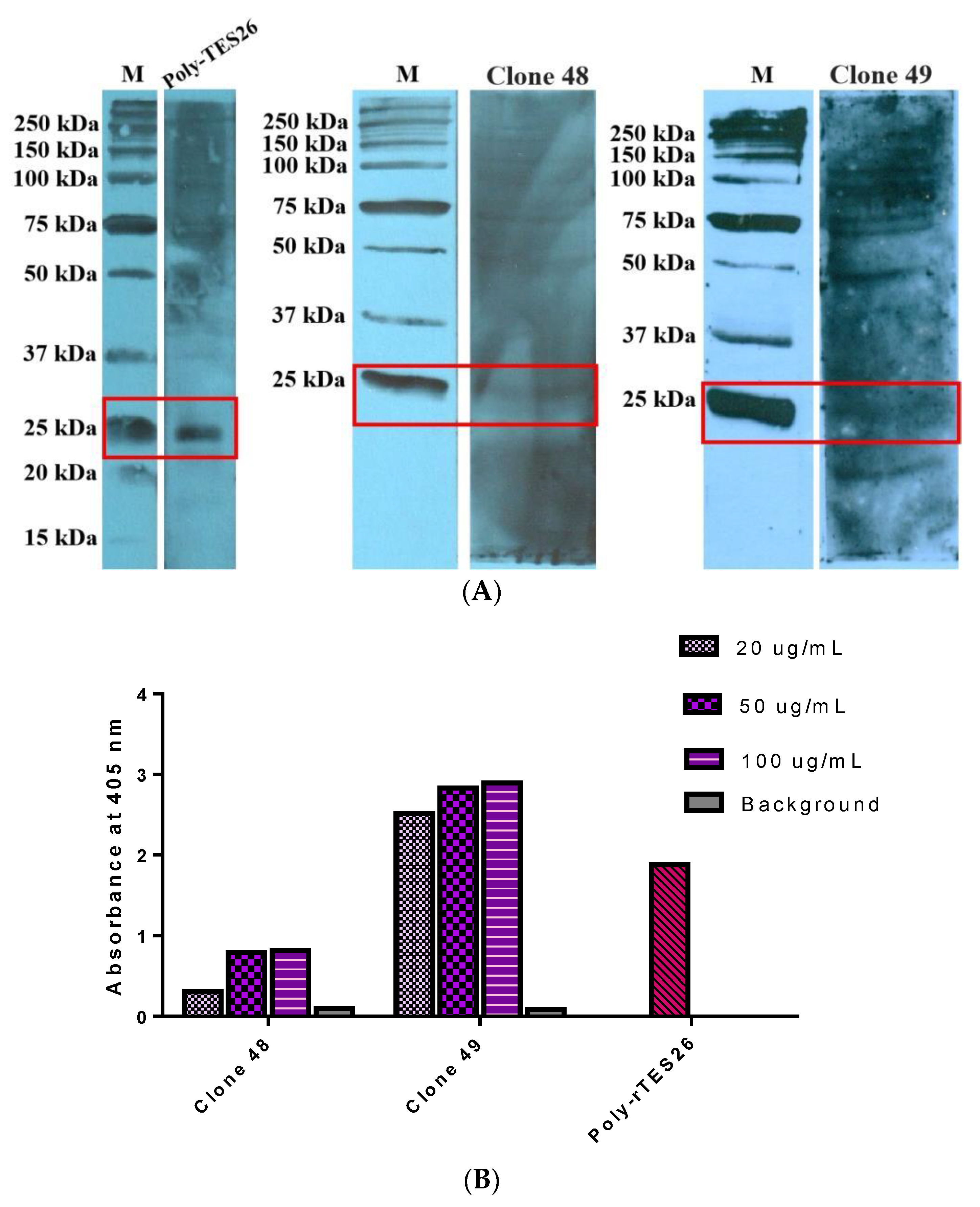
3.6. Antigen–Antibody Binding Assays
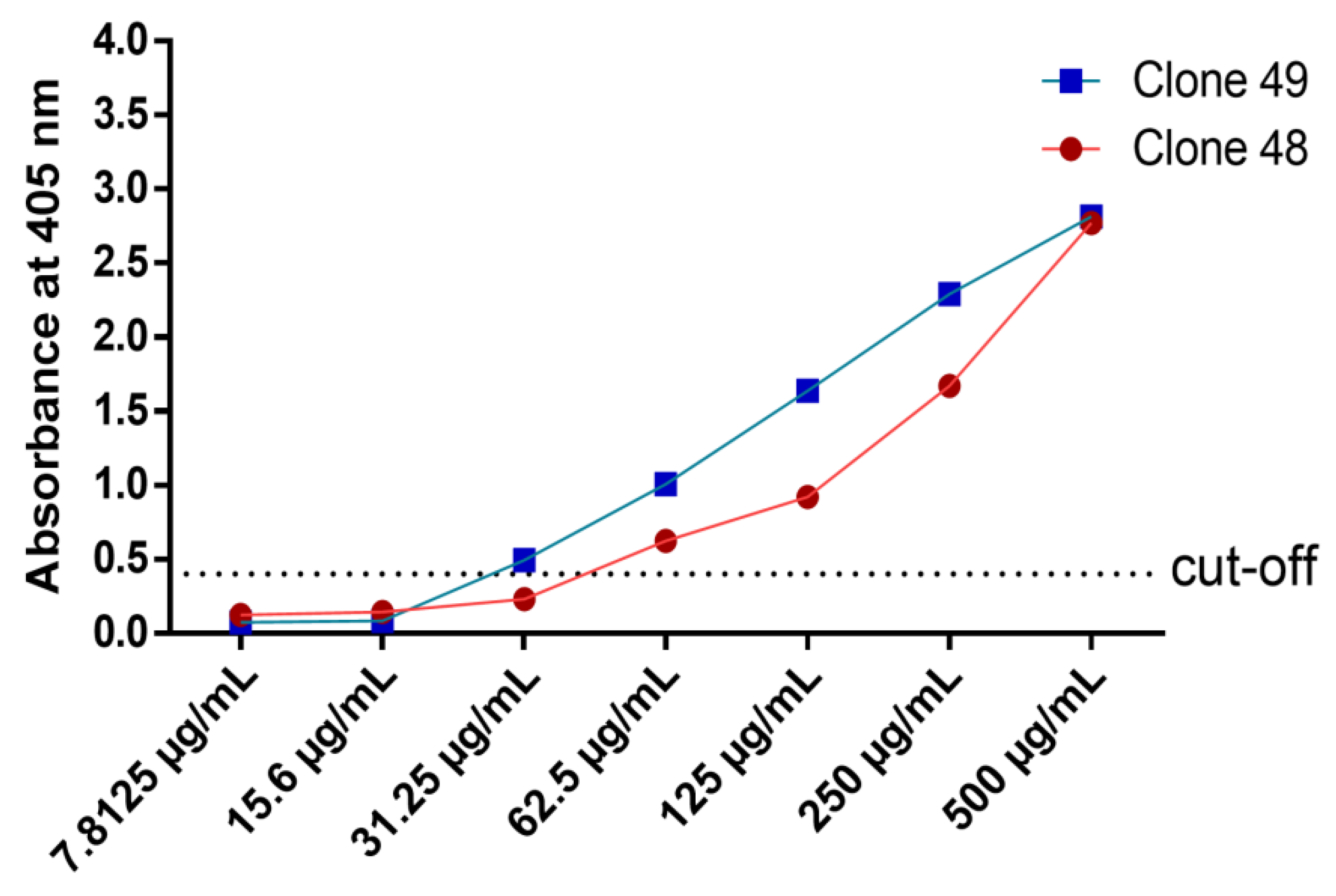
3.7. Titration ELISA
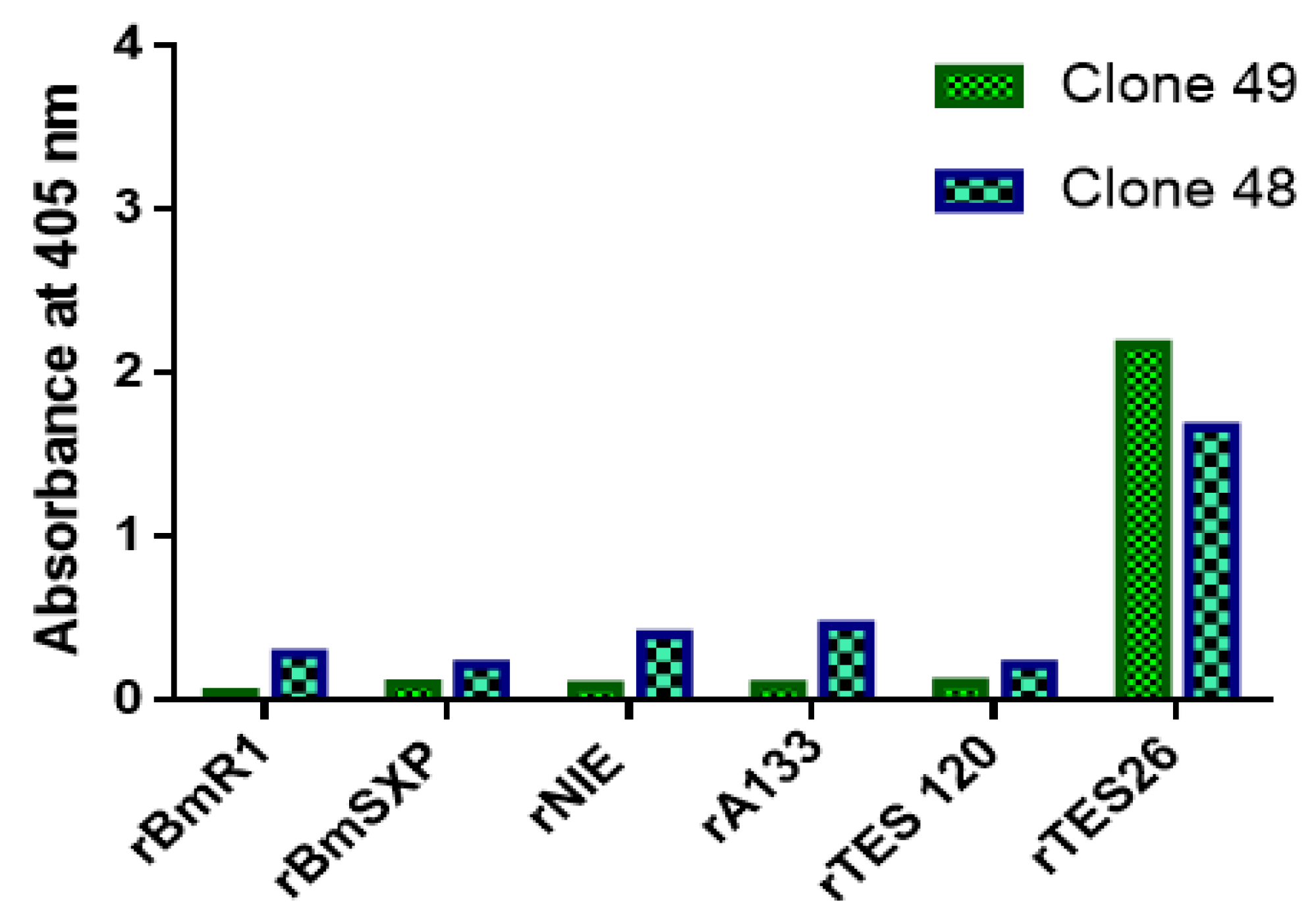
3.8. Specificity ELISA
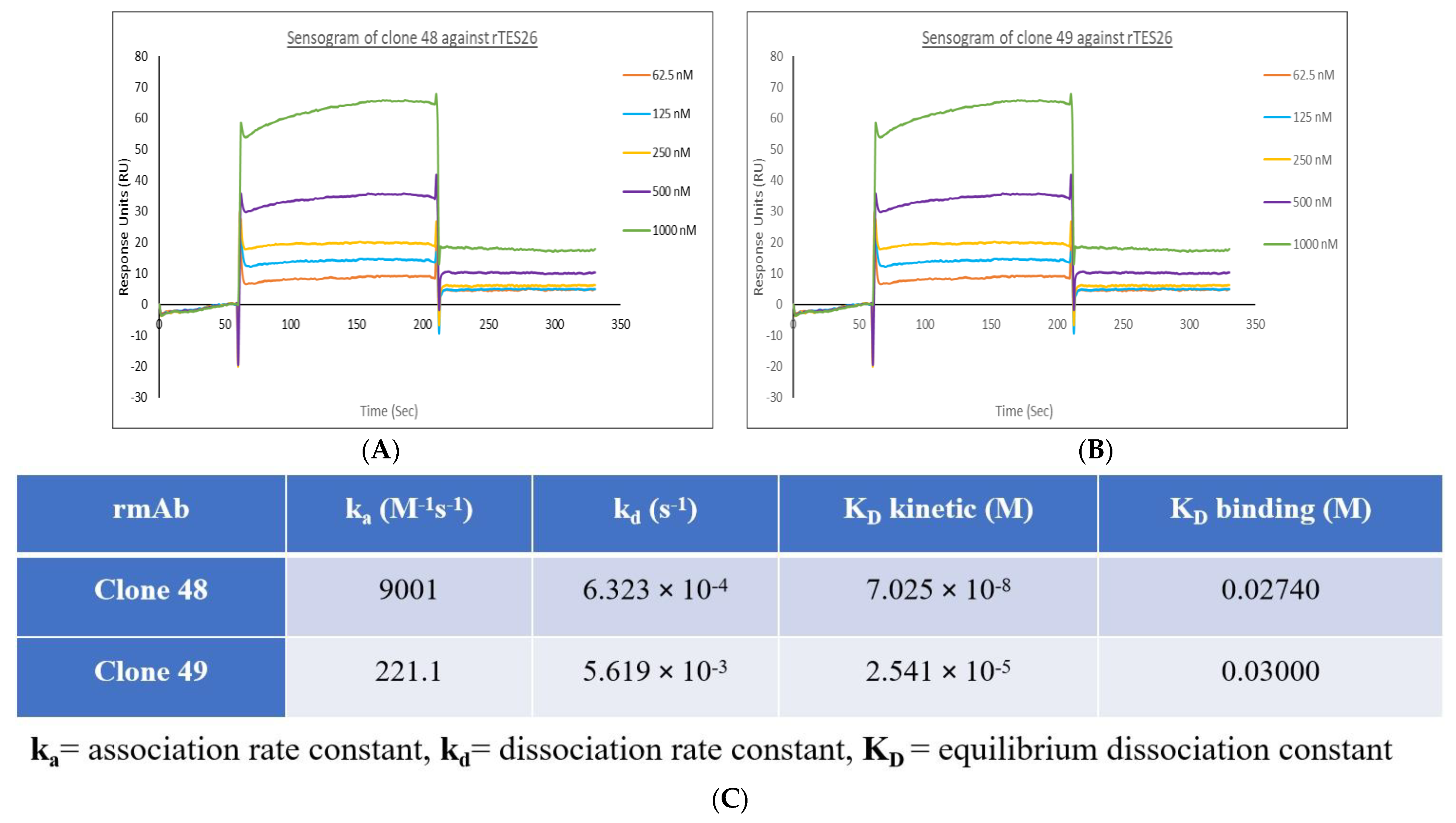
3.9. Surface Plasmon Resonance
3.10. Preliminary Antigen Detection ELISA Using Human Serum Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rostami, A.; Ma, G.; Wang, T.; Koehler, A.V.; Hofmann, A.; Chang, B.C.H.; Macpherson, C.N.; Gasser, R.B. Human toxocariasis—A look at a neglected disease through an epidemiological ‘prism’. Infect. Genet. Evol. 2019, 74, 104002. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; Liu, G.H.; Zheng, W.B.; Hong, S.J.; Sugiyama, H.; Zhu, X.Q.; Elsheikha, H.M. Toxocariasis: A silent threat with a progressive public health impact. Infect. Dis. Poverty 2018, 7, 59. [Google Scholar] [CrossRef]
- Rostami, A.; Riahi, S.M.; Holland, C.V.; Taghipour, A.; Khalili-Fomeshi, M.; Fakhri, Y.; Omrani, V.F.; Hotez, P.J.; Gasser, R.B. Seroprevalence estimates for toxocariasis in people worldwide: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2019, 13, e0007809. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, C.N.L. The epidemiology and public health importance of toxocariasis: A zoonosis of global importance. Int. J. Parasitol. 2013, 43, 999–1008. [Google Scholar] [CrossRef]
- Ma, G.; Holland, C.V.; Wang, T.; Hofmann, A.; Fan, C.K.; Maizels, R.M.; Hotez, P.J.; Gasser, R.B. Human toxocariasis. Lancet Infect. Dis. 2018, 18, e14–e24. [Google Scholar] [CrossRef]
- Overgaauw, P.A.M.; Knapen, F.V. Veterinary Parasitology Veterinary and public health aspects of Toxocara spp. Vet. Parasitol. 2013, 193, 398–403. [Google Scholar] [CrossRef]
- Auer, H.; Walochnik, J. Toxocariasis and the clinical spectrum. Adv. Parasitol. 2020, 109, 111–130. [Google Scholar] [CrossRef]
- Noordin, R.; Yunus, M.H.; Tan Farrizam, S.N.; Arifin, N. Serodiagnostic methods for diagnosing larval toxocariasis. Adv. Parasitol. 2020, 109, 131–152. [Google Scholar] [CrossRef]
- Smith, H.; Noordin, R. Diagnostic Limitations and Future Trends in the Serodiagnosis of Human Toxocariasis. In Toxcara the Enigmatic Parasite; Holland, C.V., Smith, H.V., Eds.; CABI Publishing: Wallingford, UK, 2006; pp. 93–102. [Google Scholar]
- Mohamad, S.; Azmi, N.C.; Noordin, R. Development and evaluation of a sensitive and specific assay for diagnosis of human toxocariasis by use of three recombinant antigens (TES-26, TES-30USM, and TES-120). J. Clin. Microbiol. 2009, 47, 1712–1717. [Google Scholar] [CrossRef]
- Wickramasinghe, S.; Yatawara, L.; Nagataki, M.; Takamoto, M.; Watanabe, Y.; Rajapakse, R.P.V.J.; Uda, K.; Suzuki, T.; Agatsuma, T. Development of a highly sensitive IgG-ELISA based on recombinant arginine kinase of Toxocara canis for serodiagnosis of visceral larva migrans in the murine model. Parasitol. Res. 2008, 103, 853–858. [Google Scholar] [CrossRef]
- Yamasaki, H.; Araki, K.; Lim, P.K.C.; Zasmy, N.; Mak, J.W.; Taib, R.; Aoki, T. Development of a highly specific recombinant Toxocara canis second-stage larva excretory-secretory antigen for immunodiagnosis of human toxocariasis. J. Clin. Microbiol. 2000, 38, 1409–1413. [Google Scholar] [CrossRef] [PubMed]
- Yunus, M.H.; Farrizam, S.N.T.; Karim, Z.A.; Noordin, R. A lateral flow rapid test for human toxocariasis developed using three toxocara canis recombinant antigens. Am. J. Trop. Med. Hyg. 2018, 98, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Holland, C.; Taylor, M.; Magnaval, J.F.; Schantz, P.; Maizels, R. How common is human toxocariasis? Towards standardizing our knowledge. Trends Parasitol. 2009, 25, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Magnaval, J.F.; Bouhsira, E.; Fillaux, J. Therapy and Prevention for Human Toxocariasis. Microorganisms 2022, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.H.; Bidwell, B.; Voller, A.; Robertson, B.D.; Maizels, R.M. Diagnosis of human toxocariasis by antigen capture enzyme linked immunosorbent assay. J. Clin. Pathol. 1993, 46, 551–554. [Google Scholar] [CrossRef]
- Robertson, B.D.; Burkot, T.R.; Gillespie, S.H.; Kennedy, M.W.; Wambai, Z.; Maizels, R.M. Detection of circulating parasite antigen and specific antibody in Toxocara canis infections. Clin. Exp. Immunol. 1988, 74, 236–241. [Google Scholar]
- Ishiyamna, S.; Ono, K.; Rai, S.K.; Uga, S. Method for detecting circulating Toxocara canis antigen and its application in human serum samples. Nepal Med. Coll. J. 2009, 11, 9–13. [Google Scholar]
- Zibaei, M.; Sadjjadi, S.M.; Ishiyama, S.; Sarkari, B.; Uga, S. Production of monoclonal antibody against toxocara cati second-stage larvae and its application for the detection of circulating antigens. Hybridoma 2010, 29, 217–220. [Google Scholar] [CrossRef]
- Lelli, D.; Moreno, A.; Brocchi, E.; Sozzi, E.; Capucci, L.; Canelli, E.; Barbieri, I.; Zeller, H.; Cordioli, P. West Nile virus: Characterization and diagnostic applications of monoclonal antibodies. Virol. J. 2012, 9, 1–11. [Google Scholar] [CrossRef]
- Varghese, A.; Raina, O.K.; Chandra, D.; Mirdha, B.R.; Kelawala, N.H.; Solanki, J.B.; Kumar, N.; Ravindran, R.; Arun, A.; Rialch, A.; et al. Sero-detection of Toxocara canis infection in human with T.canis recombinant arginine kinase, cathepsin L-1 and TES-26 antigens. Acta Parasitol. 2017, 62, 775–778. [Google Scholar] [CrossRef]
- Rahumatullah, A.; Ahmad, A.; Noordin, R.; Lim, T.S. Delineation of BmSXP antibody V-gene usage from a lymphatic filariasis based immune scFv antibody library. Mol. Immunol. 2015, 67, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Rahumatullah, A.; Karim, I.Z.A.; Noordin, R.; Lim, T.S. Antibody-based protective immunity against helminth infections: Antibody phage display derived antibodies against BmR1 antigen. Int. J. Mol. Sci. 2017, 18, 2376. [Google Scholar] [CrossRef] [PubMed]
- Rahumatullah, A.; Yunus, M.H.; Tye, G.J.; Noordin, R. Applications of recombinant monoclonal antibodies against filarial antigen proteins. Am. J. Trop. Med. Hyg. 2020, 102, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Rahumatullah, A.; Balachandra, D.; Noordin, R.; Baharudeen, Z.; Lim, Y.Y.; Chong, Y.S.; Lim, T.S. Broad specificity of immune helminth scFv library to identify monoclonal antibodies targeting Strongyloides. Sci. Rep. 2021, 11, 2502. [Google Scholar] [CrossRef] [PubMed]
- Retter, I.; Althaus, H.H.; Münch, R.; Müller, W. VBASE2, an integrative V gene database. Nucleic Acids Res. 2005, 33, 671–674. [Google Scholar] [CrossRef]
- Brochet, X.; Lefranc, M.P.; Giudicelli, V. IMGT/V-QUEST: The highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008, 36, 503–508. [Google Scholar] [CrossRef]
- Löfås, S.; Johnsson, B. A novel hydrogel matrix on gold surfaces in surface plasmon resonance sensors for fast and efficient covalent immobilization of ligands. J. Chem. Soc. Chem. Commun. 1990, 21, 1526–1528. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef]
- Schasfoort, R.B.M. Introduction to Surface Plasmon Resonance. In Handbook Surface Plasmon Resonance, 2nd ed.; Schasfoort, R.B.M., Ed.; Royal Society of Chemistry: London, UK, 2017; pp. 1–26. [Google Scholar] [CrossRef]
- Siddiqui, M.Z. Monoclonal antibodies as diagnostics; an appraisal. Indian J. Pharm. Sci. 2010, 72, 12. [Google Scholar] [CrossRef]
- Jaffe, C.L.; Bennett, E.; Grimaldi, G.; McMahon-Pratt, D. Production and characterization of species-specific monoclonal antibodies against Leishmania donovani for immunodiagnosis. J. Immunol. 1984, 133, 440–447. [Google Scholar]
- Parish, N.M.; Morrison, W.I.; Pearson, T.W. Identification of an antigen specific to Trypanosoma congolense by using monoclonal antibodies. J. Immunol. 1985, 134, 593–597. [Google Scholar]
- Wright, I.G.; White, M.; Tracey-Patte, P.D.; Donaldson, R.A.; Goodger, B.V.; Waltisbuhl, D.J.; Mahoney, D.F. Babesia bovis: Isolation of a protective antigen by using monoclonal antibodies. Infect. Immun. 1983, 41, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Hirose, H.; Matsuda, H.; Murata, M.; Sekiya, Y. Preparation of monoclonal antibodies against Marek’s disease virus and herpesvirus of turkeys. Jpn. J. Vet. Sci. 1986, 48, 1263–1266. [Google Scholar] [CrossRef] [PubMed]
- Lyaku, J.R.; Sinclair, J.A.; Nettleton, P.F.; Marsden, H.S. Production and characterization of monoclonal antibodies to cervine herpesvirus-1. Vet. Microbiol. 1992, 32, 229–239. [Google Scholar] [PubMed]
- Marchioli, C.; Yancey, R.J., Jr.; Timmins, J.G.; Post, L.E.; Young, B.R.; Povendo, D.A. Protection of mice and swine from pseudorabies virus-induced mortality by administration of pseudorabies virus-specific mouse monoclonal antibodies. Am. J. Vet. Res. 1988, 49, 860–864. [Google Scholar] [PubMed]
- Lim, B.N.; Tye, G.J.; Choong, Y.S.; Ong, E.B.B.; Ismail, A.; Lim, T.S. Principles and application of antibody libraries for infectious diseases. Biotechnol. Lett. 2014, 36, 2381–2392. [Google Scholar] [CrossRef]
- Anderson, J.P.; Rascoe, L.N.; Levert, K.; Chastain, H.M.; Reed, M.S.; Rivera, H.N.; McAuliffe, I.; Zhan, B.; Wiegand, R.E.; Hotez, P.J. Development of a Luminex bead based assay for diagnosis of toxocariasis using recombinant antigens Tc-CTL-1 and Tc-TES-26. PLoS Negl. Trop. Dis. 2015, 9, e0004168. [Google Scholar] [CrossRef]
- Mole, C.M.; Bene, M.C.; Montagne, P.M.; Seilles, E.; Faure, G.C. Light chains of immunoglobulins in human secretions. Clin. Chim. Acta 1994, 224, 191–197. [Google Scholar] [CrossRef]
- Montaño, R.F.; Morrison, S.L. Influence of the isotype of the light chain on the properties of IgG. J. Immunol. 2002, 168, 224–231. [Google Scholar] [CrossRef]
- Hust, M.; Meyer, T.; Voedisch, B.; Rulker, T.; Thie, H.; El-Ghezal, A.; Kirsch, M.I.; Schutte, M.; Helmsing, S.; Meier, D. A human scFv antibody generation pipeline for proteome research. J. Biotechnol. 2011, 152, 159–170. [Google Scholar] [CrossRef]
- Lloyd, C.; Lowe, D.; Edwards, B.; Welsh, F.; Dilks, T.; Hardman, C.; Vaughan, T. Modelling the human immune response: Performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 2009, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.D.J.; Clarke, K.F.; Conquer, J.S.; Crofts, A.M.; Crowther, S.R.E.; Dyson, M.R. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8, R254. [Google Scholar] [CrossRef] [PubMed]
- Sajadi, M.M.; Farshidpour, M.; Brown, E.P.; Ouyang, X.; Seaman, M.S.; Pazgier, M.; Ackerman, M.E.; Robinson, H.; Tomaras, G.; Parsons, M.S.; et al. λ Light Chain Bias Associated With Enhanced Binding and Function of Anti-HIV Env Glycoprotein Antibodies. J. Infect. Dis. 2016, 213, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; To, R.; Kandalaft, H.; Ding, W.; van Faassen, H.; Luo, Y.; Schrag, J.D.; St-Amant, N.; Hefford, M.; Hirama, T. Antibody light chain variable domains and their biophysically improved versions for human immunotherapy. MAbs 2014, 6, 219–235. [Google Scholar] [CrossRef][Green Version]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003, 60, 523–533. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
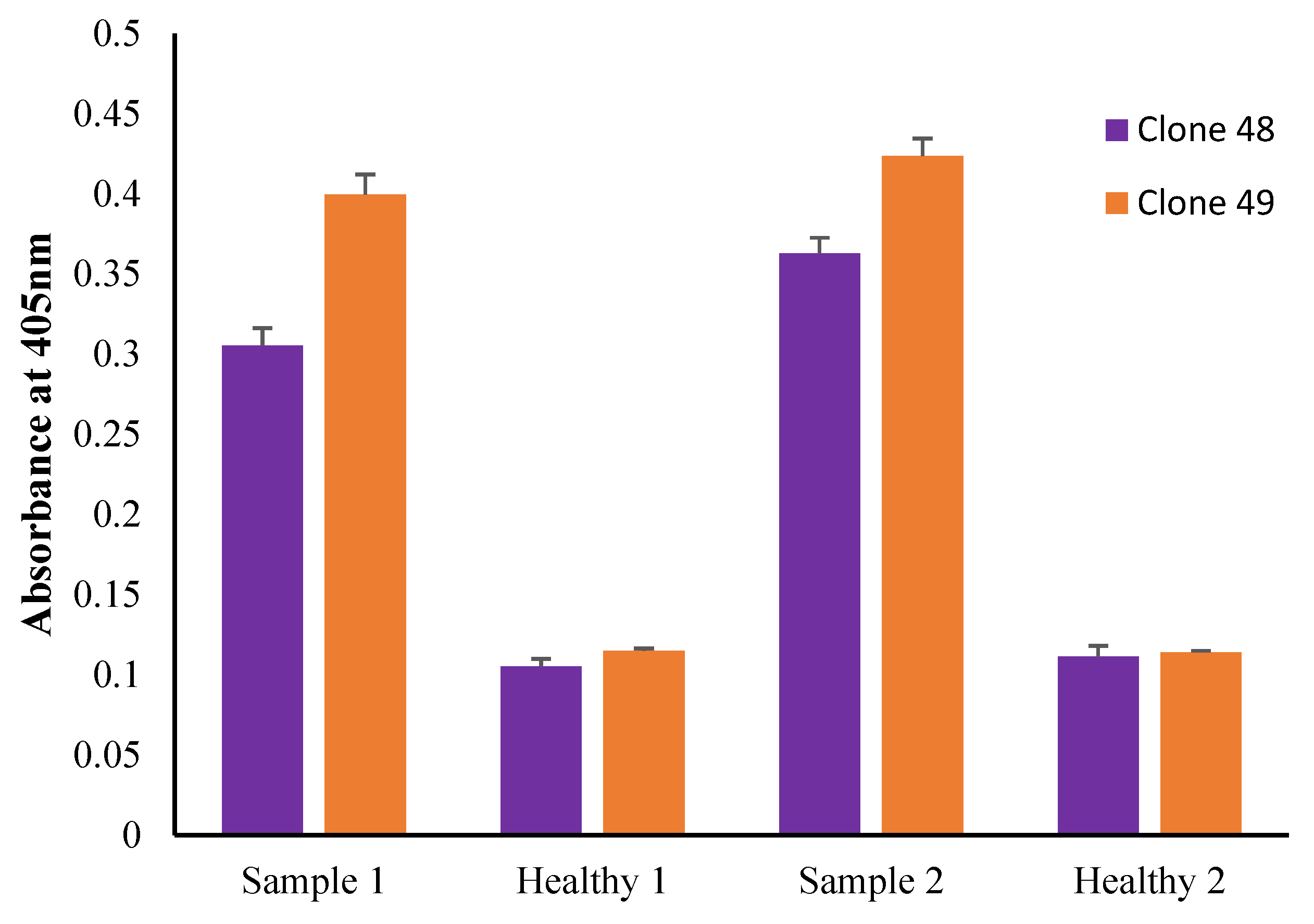{kind=link}
| Clone Name | Gene Family | |
|---|---|---|
| Heavy Chain (VH) | Light Chain (VL) | |
| Clone 22 | IgVH3 | IgVL1 |
| Clone 48 | IgVH3 | IgVL1 |
| Clone 49 | IgVH3 | IgVL3 |
| Clone 50 | IgVH6 | IgVL3 |
| Clone 51 | No VH | IgVL3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baharudeen, Z.; Noordin, R.; Soon, L.T.; Balachandra, D.; Anuar, N.S.; Mustafa, F.H.; Rahumatullah, A. Isolation and Production of Human Monoclonal Antibody Proteins against a Toxocara canis Excretory–Secretory Recombinant Antigen. Pathogens 2022, 11, 1232. https://doi.org/10.3390/pathogens11111232
Baharudeen Z, Noordin R, Soon LT, Balachandra D, Anuar NS, Mustafa FH, Rahumatullah A. Isolation and Production of Human Monoclonal Antibody Proteins against a Toxocara canis Excretory–Secretory Recombinant Antigen. Pathogens. 2022; 11(11):1232. https://doi.org/10.3390/pathogens11111232
Chicago/Turabian StyleBaharudeen, Zamrina, Rahmah Noordin, Lim Theam Soon, Dinesh Balachandra, Nor Suhada Anuar, Fatin Hamimi Mustafa, and Anizah Rahumatullah. 2022. "Isolation and Production of Human Monoclonal Antibody Proteins against a Toxocara canis Excretory–Secretory Recombinant Antigen" Pathogens 11, no. 11: 1232. https://doi.org/10.3390/pathogens11111232
APA StyleBaharudeen, Z., Noordin, R., Soon, L. T., Balachandra, D., Anuar, N. S., Mustafa, F. H., & Rahumatullah, A. (2022). Isolation and Production of Human Monoclonal Antibody Proteins against a Toxocara canis Excretory–Secretory Recombinant Antigen. Pathogens, 11(11), 1232. https://doi.org/10.3390/pathogens11111232