
Genetic Diversity of Venturia inaequalis in Latvia Revealed by Microsatellite Markers


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Fungal Isolation
2.2. DNA Extraction
2.3. PCR Amplification
2.4. Data Analysis
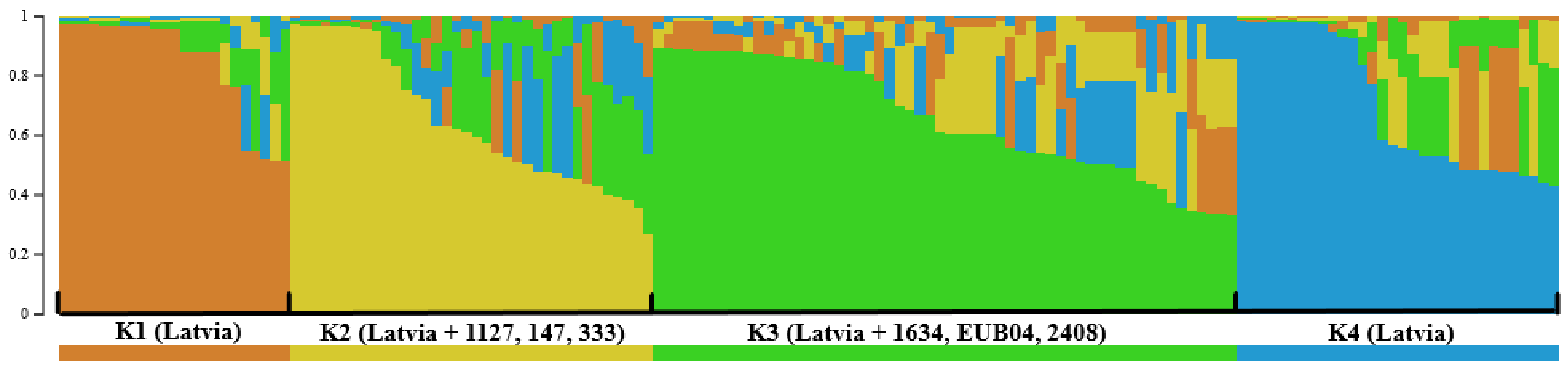
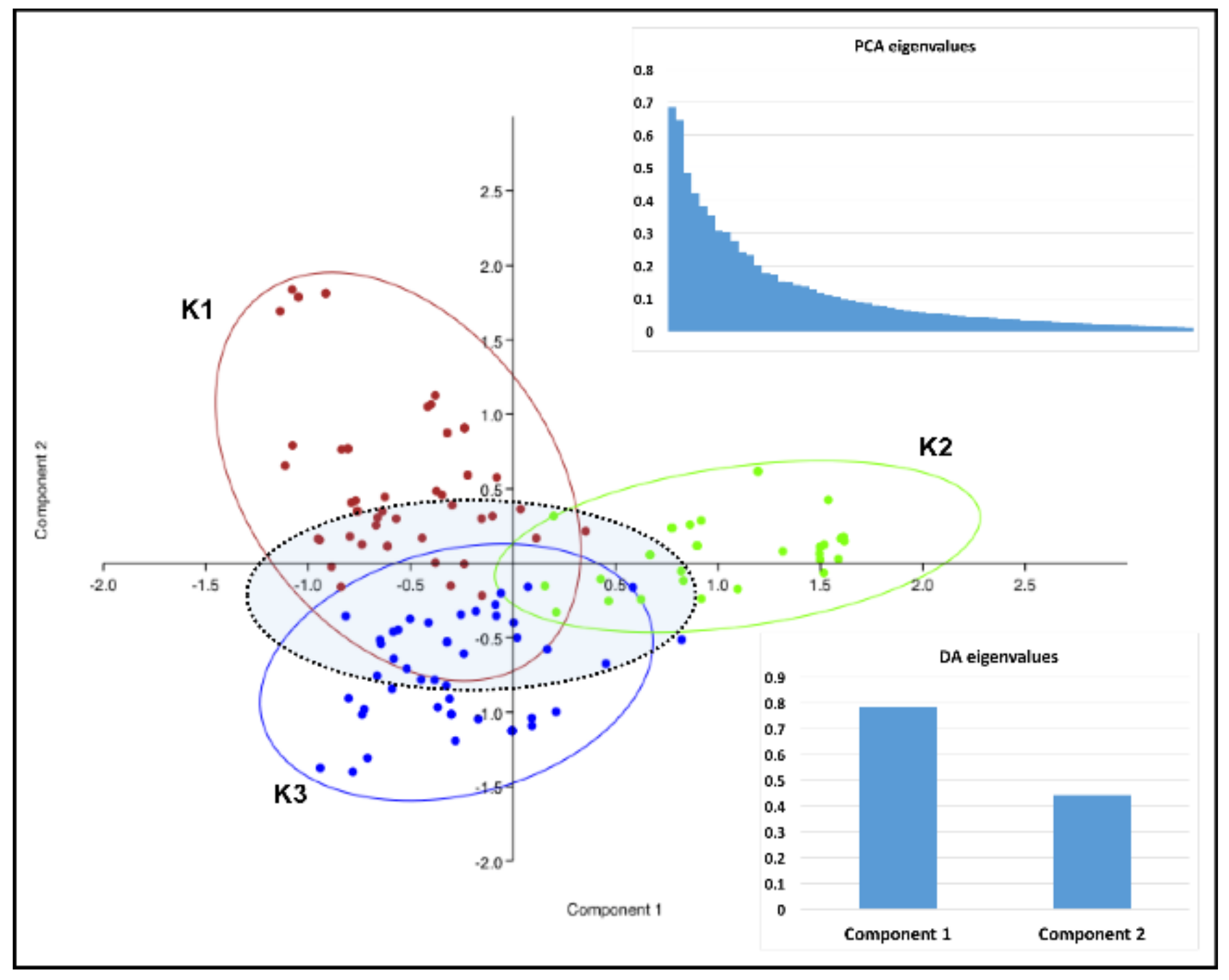
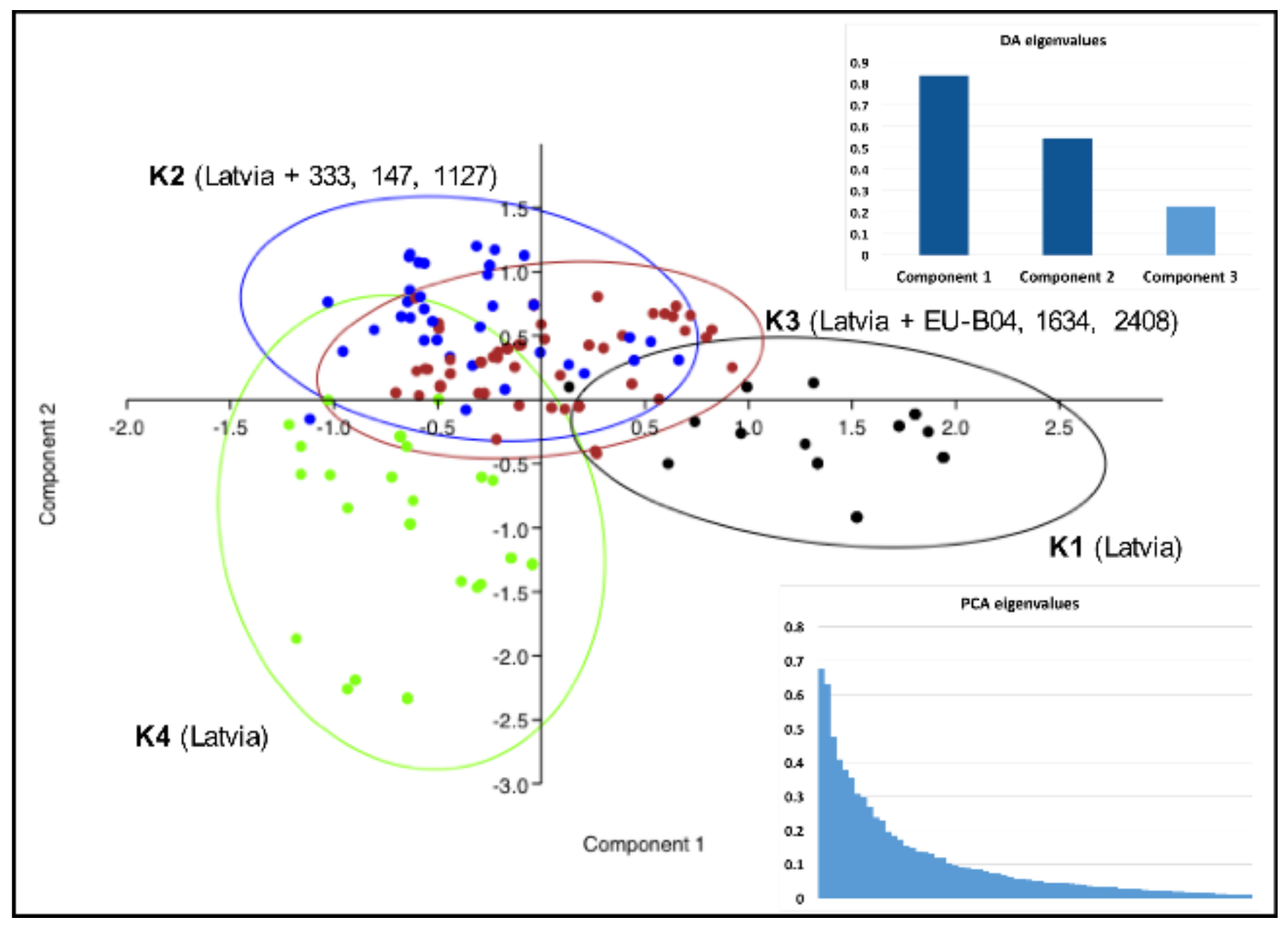
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MacHardy, W. Inheritance of resistance to Venturia inaequalis. In Apple Scab, Biology, Epidemiology and Management; APS: St. Paul, MN, USA, 1996; pp. 61–103. [Google Scholar]
- Suprun, I.I.; Nasonov, A.I.; Tokmakov, S.V.; Barsukova, O.N.; Yakuba, G.V. Application of SSR markers for study of genetic diversity of Venturia inaequalis in the different types of orchards in the north caucasian region. Agric. Biol. 2018, 53, 170–178. [Google Scholar]
- Soriano, J.M.; Joshi, S.G.; Van Kaauwen, M.; Noordijk, Y.; Groenwold, R.; Henken, B.; Van de Weg, W.E.; Schouten, H.J. Identification and mapping of the novel apple scab resistance gene Vd3. Tree Genet. Genomes. 2009, 5, 475–482. [Google Scholar] [CrossRef]
- Jamar, L. Innovative Strategies for the Control of Apple Scab (Venturia inaequalis [Cke.] Wint.) in Organic Apple Production. Ph.D. Thesis, University of Liege-Gembloux Agro-Bio Tech, Gembloux, Belgium, 2011. [Google Scholar]
- Ellis, M.A.; Ferre, D.C.; Madden, L.V. Effects of an apple scab-resistant cultivar on use patterns of inorganic and organic fungicides and economics of disease control. Plant Dis. 1998, 82, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Corroyer, N.; Petit, J.L. Le pommier. In Dans: Produire des Fruits en Agriculture Biologique; ITAB: Paris, France, 2002; pp. 230–261. [Google Scholar]
- Eihe, M.; Rancane, R.; Vilka, L. Different fungicide combinationsagainst apple scab helping to avoid fungus resistance. Sodininkystë ir Darþininkystë. 2009, 28, 57–67. [Google Scholar]
- Grantiņa-Ieviņa, L.; Rancāne, R.; Jakobija, I.; Ērgle, G. The distribution of apple scab on the most widely grown apple varieties in different regions of Latvia. In Proceedings of the Ābeļu Kraupja izplatība uz Plašāk Audzētajām Ābeļu Šķirnēm Dažādos Latvijas Reģionos. Rakstu Krājums Vecauce—2015: Lauksaimniecības Zinātne Reorganizācijas Laikā; LLU, Vecauce, Latvia, 5 November 2015; pp. 25–28. (In Latvian). [Google Scholar]
- Ebrahimi, L.; Fotouhifar, K.-B.; Javan Nikkhah, M.; Naghavi, M.-R.; Baisakh, N. Population Genetic Structure of Apple Scab (Venturia inaequalis (Cooke)G. Winter) in Iran. PLoS ONE 2016, 11(9), e0160737. [Google Scholar] [CrossRef]
- Bus, V.G.M.; Rikkerink, E.H.A.; Caffier, V.; Durel, C.E.; Plummer, K.M. Revision of the nomenclature of the differential host—pathogen interactions of Venturia inaequalis and Malus. Annu. Rev. Phytopathol. 2011, 49, 391–413. [Google Scholar] [CrossRef]
- Patocchi, A.; Wehrli, A.; Dubuis, P.-H.; Auwerkerken, A.; Leida, C.; Cipriani, G.; Passey, T.; Staples, M.; Didelot, F.; Philion, V.; et al. Ten years of VINQUEST: First insight for breeding new apple cultivars with durable apple scab resistance. Plant. Dis. 2020, 104, 2074–2081. [Google Scholar] [CrossRef]
- Tenzer, I.; Gessler, C. Subdivision and genetic structure of four populations of Venturia inaequalis in Switzerland. Eur. J. Plant Pathol. 1997, 103, 565–571. [Google Scholar] [CrossRef]
- Tenzer, I.; Gessler, C. Genetic diversity of Venturia inaequalis across Europe. Eur. J. Plant Pathol. 1999, 105, 545–552. [Google Scholar] [CrossRef]
- Xu, X.; Yang, J.; Thakur, V.; Roberts, A.; Barbara, D.J. Population variation of apple scab (Venturia inaequalis) isolates from Asia and Europe. Plant Dis. 2008, 92, 247–252. [Google Scholar] [CrossRef]
- Boehm, E.W.A.; Freeman, S.; Shabi, E.; Michailides, T.J. Microsatellite primers indicatet hepresence of asexual populations of Venturia inaequalis in coastal Israeli apple orchards. Phytoparasitica 2003, 31, 236–251. [Google Scholar] [CrossRef]
- Gladieux, P.; Zhang, X.G.; Afoufa-Bastien, D.; Valdebenito Sanhueza, R.M.; Sbaghi, M.; Le Cam, B. On the origin and spread of the scab disease of apple: Out of central Asia. PLoS ONE 2008, 3, 1455. [Google Scholar] [CrossRef] [PubMed]
- Kaymak, S.; Boyraz, N.; Daniels, J. Molecular markers to evaluate genetic diversity among Venturia inaequalis isolates obtained from apple plantations in Isparta Province. Turk. J. Agric. For. 2016, 40, 489–498. [Google Scholar] [CrossRef]
- Koopman, T.A.; Meitz-Hopkins, J.C.; Bester-van der Merwe, A.E.; Tobutt, K.R.; Bester, C.; Lennox, C.L. Genetic diversity and gene flow of four South African Venturia inaequalis (apple scab) populations. Phytopathology 2017, 107, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Sitther, V.; Garrido Haro, P.A.; Molineros, J.E.; Garzon, C.D.; Jimenez-Gasco, M.M. Genetic diversity ofapple- and crabapple-infecting isolates of Venturia inaequalis in Pennsylvania, the United States, deter-mined by microsatellite markers. For. Pathol. 2018, 48, 1–9. [Google Scholar] [CrossRef]
- Mezhnina, O.A.; Urbanovich, O.Y.; Gashenko, T.A.; Kozlovskaya, Z.A. The study of the genetic diversity of the Belarusian population Venturia inaequalis Cooke (Wint.) using SSR-markers. In Biotekhnologicheskie Priemy v Sokhranenii Bioraznoobraziia i Selektsii Rasteniy: Sbornik Statei Mezh-Dunarodnoi Nauchnoi Konferentsii Minsk. In Proceedings of the International Scientific Conference (Сбoрник статей Междунарoднoй научнoй кoнференции), Minsk, Belarus, 18–20 August; Beloarus, Minsk, 2014; pp. 168–171. (In Russian). [Google Scholar]
- Guérin, F.; Franck, P.; Loiseau, A.; Devaux, M.; Le Cam, B. Isolation of 21 new polymorphic microsatellite loci in the phytopathogenic fungus Venturia inaequalis. Mol. Ecol. 2004, 4, 268–270. [Google Scholar] [CrossRef]
- Xu, X.; Harvey, N.; Roberts, A.; Barbara, D. Population variation of apple scab (Venturia inaequalis) within mixed orchards in the UK. Eur. J. Plant Pathol. 2013, 135, 97–104. [Google Scholar] [CrossRef]
- Mansoor, S.; Ahmed, N.; Sharma, V.; Jan, S.; Nabi, S.U.; Mir, J.I.; Mir, M.A.; Masoodi, K.Z. Elucidating genetic variability and population structure in Venturia inaequalis associated with apple scab disease using SSR markers. PLoS ONE 2019, 14, 1–16. [Google Scholar] [CrossRef]
- Papp, D.; Singh, J.; Gadoury, D.; Khan, A. New North American isolates of Venturia inaequalis can overcome apple scab resistance of Malus floribunda 821. Plant Dis. 2020, 104, 649–655. [Google Scholar] [CrossRef]
- Morgante, M.; Olivieri, A.M. PCR-amplified microsatellites as markers in plant genetics. Plant J. 1993, 3, 175–182. [Google Scholar] [CrossRef]
- Ikase, L.; Feldmane, D.; Rubauskis, E.; Skrīvele, M.; Strautiņa, S.; Drudze, I.; Grāvīte, I.; Juhņēviča-Radenkova, K.; Kalniņa, I.; Kaufmane, E.; et al. Augļkopība; LV, Augļkopības Institūts: Dobele, Latvia, 2015; p. 544. (In Latvian) [Google Scholar]
- Didelot, F.; Brun, L.; Parisi, L. Effects of cultivar mixtures on scab control in apple orchards. Plant Pathol. 2007, 56, 1014–1022. [Google Scholar] [CrossRef]
- Melounová, M.; Vejl, P.; Sedlák, P.; Reznerová, A.; Tesařová, M.; Blažek, J.; Zoufalá, J. The variability of Venturia inaequalis CKE. races in the Czech Republic and the accumulation of resistance genes in apple germplasm. Plant Soil Environ. 2004, 50, 416–423. [Google Scholar] [CrossRef]
- Dhingra, O.D.; Sinclair, J.B. Basic Plant Pathological Methods; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Caffier, V.; Patocchi, A.; Expert, P.; Bellanger, M.-N.; Durel, C.-E.; Bodmer, M.H.; Broggini, G.A.L.; Groenwold, R.; Bus, V.G.M. Virulence characterization of Venturia inaequalis reference isolates on the differential set of Malus hosts. Plant Dis. 2015, 99, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, I.; Ivanissevich, S.D.; Morgante, M.; Gessler, C. Identification of microsatellite markers and their application to population genetics of Venturia inaequalis. Phytopathology 1999, 89, 748–753. [Google Scholar] [CrossRef]
- Xu, X.; Roberts, T.; Barbara, D.; Harvey, N.G.; Gao, L.; Sargent, D.J. A genetic linkage map of Venturia inaequalis, the causal agent of apple scab. BMC Res. Notes 2009, 2, 1–6. [Google Scholar] [CrossRef]
- Lamboy, W.F.; Alpha, C.G. Using simple sequence repeats (SSRs) for DNA fingerprinting germplasm accessions of grape (Vitis L.) species. J. Amer. Soc. Hort. Sci. 1998, 123, 182–188. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for educationand data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Pritchard, J.K.; Stephans, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing withlabel switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.K.; Ramasamy, S.; Bindroo, B.B.; Naik, V.G. STRUCTURE PLOT: A program for drawing elegant STRUCTURE bar plots in user friendly interface. SpringerPlus 2014, 3, 431. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.A.; Linde, C. Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 2002, 40, 349–379. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tao, F.; Fan, S.; Li, H.; Yang, J.; Gao, L. Genetic diversity of Venturia inaequalis isolates (Apple scab) in China and U.K. determined by SSR markers. PLoS ONE 2021, 16, e0252865. [Google Scholar] [CrossRef]
- Guérin, F.; Gladieux, P.; Le Cam, B. Origin and colonization history of newly virulent strains of the phyto-pathogenic fungus Venturia inaequalis. Fungal Genet. Biol. 2007, 44, 284–292. [Google Scholar] [CrossRef]
- Clark, M.D. Characterizing the Host Response and Genetic Control of Resistance in ‘Honeycrisp’ to Apple Scab (Venturia inaequalis). Ph.D. Dissertation, University of Minnesota, Saint Paul, MN, USA, 2014. Available online: http://conservancy.umn.edu/handle/11299/172140 (accessed on 20 May 2022).
- Sokolova, O.; Moročko-Bičevska, I. Evaluation of apple scab and occurrence of Venturia inaequalis races on differential Malus genotypes in Latvia. Proc. Latv. Acad. Sci. Sect. B 2022, in press. [CrossRef]
- Pikunova, A.V.; Sedov, E.N. The composition of Venturia inaequalis races in the Oryol region [Расoвый сoстав Venturia inaequalis в услoвиях Орлoвскoй oбласти]. Mycol. Phytopathol. 2019, 57, 293–300. (In Russian) [Google Scholar] [CrossRef]
- Leroy, T.; Lemaire, C.; Dunemann, F.; Cam, B.L. The genetic structure of a Venturia inaequalis population composed of different Malus species. BMC Evol. Biol. 2013, 13, 64. [Google Scholar] [CrossRef]
- Rancane, R.; Ozoliņa-Pole, L. Observations of the LLU Plant Protection Scientific Institute “Agrihorts” on the spread of harmful organisms in the apple orchards in the 2021 season. [LLU Augu aizsardzības zinātniskā institūta “Agrihorts” novērojumi par kaitīgo organismu izplatību ābeļu stādījumos 2021. gada sezonā]. Prof. Hortic. 2021, 1, 49–53. (In Latvian) [Google Scholar]
- Kaufmane, E.; Skrivele, M.; Rubauskis, E.; Strautiņa, S.; Ikase, L.; Lacis, G.; Priekule, I. Development of fruit science in Latvia. Proc. Latv. Acad. Sci. Sect. B Nat. Exact Appl. Sci. 2013, 67, 71–83. [Google Scholar] [CrossRef]
- Ikase, L.; Drudze, I.; Lācis, G. Current achievements of the Latvian apple breeding program. Proc. Latv. Acad. Sci. Sect. B 2022, in press.
- Ikase, L.; Lācis, G. Apple breeding and genetic resources in Latvia. Acta Hortic. 2013, 976, 69–74. [Google Scholar] [CrossRef]
- Zelmene, K.; Kārkliņa, K.; Ikase, L.; Lācis, G. Inheritance of apple (Malus x domestica (L.) Borkh) resistance against apple scab (Venturia inaequalis (Cooke) Wint.) in hybrid breeding material obtained by gene pyramiding. Horticulturae 2022, 8, 772. [Google Scholar] [CrossRef]
- Masny, S. Occurrence of Venturia inaequalis races in Poland able to overcome specific apple scab resistance genes. Eur. J. Plant Path. 2017, 147, 313–323. [Google Scholar] [CrossRef][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Locality and Host Genotype | Number of Localities (Number of Strains), Sprayed/Non-Sprayed/Total | ||
|---|---|---|---|
| Zemgale | Kurzeme | Total | |
| Commercial orchards | 3/0/3 (26/0/26) | 5/0/5 (44/0/44) | 8/0/8 (70/0/70) |
| ‘Lobo’ | 2/0/2 (19/0/19) | 2/0/2 (6/0/6) | 4/0/4 (25/0/25) |
| ‘Auksis’ | 1/0/1 (7/0/7) | 1/0//1 (2/0//2) | 2/0/1 (9/0/9) |
| ‘Kovalenkovskoje’ | - | 1/0/1 (7/0/7) | 1/0/1 (7/0/7) |
| ‘Belorusskoje Malinovoje’ | - | 2/0/2 (14/0/14) | 2/0/2 (14/0/14) |
| ‘Rubin’ | - | 1/0/1 (6/0/6) | 1/0/1 (6/0/6) |
| Malus domestica, unknown | - | 2/0/2 (9/0/9) | 2/0/2 (9/0/9) |
| Home gardens | 0/4/4 (0/23/23) | 0/1/1 (0/3/3) | 0/5/5 (0/26/26) |
| ‘Huvitus’ | 0/1/1 (0/11/11) | - | 0/1/1 (0/11/11) |
| ‘Sister of Liberty’ | 0/1/1 (0/1/1) | - | 0/1/1 (0/1/1) |
| ‘Antonovka’ | 0/1/1 (0/2/2) | - | 0/1/1 (0/2/2) |
| ‘Auksis’ | 0/1/1 (0/1/1) | - | 0/1/1 (0/1/1) |
| ‘Vista Bella’ | 0/1/1 (0/1/1) | - | 0/1/1 (0/1/1) |
| ‘Rudens Svītrainais’ | 0/1/1 (0/5/5) | - | 01/1 (0/5/5) |
| Malus domestica, unknown | 0/1/1 (0/2/2) | 0/1/1 (0/3/3) | 0/1/1 (0/3/3) |
| Germplasm collections | 1/1/2 (12/17/29) | - | 1/1/2 (12/17/29) |
| ‘Rāja’ | 0/1/1 (0/7/7) | - | 0/1/1 (0/7/7) |
| Nr.16-97-109 | 0/1/1 (0/5/5) | - | 0/1/1 (0/5/5) |
| ‘Belorusskoje Malinovoje’ | 0/1/1 (0/5/5) | - | 0/1/1 (0/5/5) |
| Nr.29-97-1 | 1/0/1 (6/0/6) | - | 1/0/1 (6/0/6) |
| ‘Stars’ | 1/0/1 (5/0/5) | - | 1/0/1 (5/0/5) |
| ‘Popes ābele’ | 1/0/1 (1/0/1) | - | 1/0/1 (1/0/1) |
| Greenery | 0/1/1 (0/18/18) | - | 0/1/1 (0/18/18) |
| Columnar apple, unknown | 0/1/1 (0/10/10) | - | 0/1/1 (0/10/10) |
| ‘Carnikava’ | 0/1/1 (0/4/4) | - | 0/1/1 (0/4/4) |
| Malus toringo | 0/1/1 (0/4/4) | - | 0/1/1 (0/4/4) |
| Total | 4/6/10 (38/58/96) | 5/1/6 (44/3/47) | 9/7/16 (82/61/143) |
| Locus | Primer Sequences, 5′–3′ | Allele Size Range, bp | Annealing Temperature, °C | Reference |
|---|---|---|---|---|
| Vicacg8/42 | F:(FAM-)TGTCAGCCACGCTAGAAG R:CACCGGACGAATCATGC | 196–232 | 60 | [21] |
| Vitg11/70 | F:(HEX-)GAAGAGGTTGGAGTGGTTG R:GAACCGAATCTGTACAGGAC | 184–196 | 60 | |
| Vigtg10/95 | F:(ROX-)AGGTGTTGCTGTCTTGGAG R:CGATAGTGTCATTTCCAATCC | 134–169 | 58 | |
| Vica9/152 | F:(FAM-)GCACCTGCTCTGTCTATCTC R:AAGGTTCAGGCACTGGAG | 167–191 | 58 | |
| Vitc2/D | F:(TAMRA-)GCTCCTTCTGGGTAAGA R:CTCTACATCTCATCCCATC | 184–278 | 58 | |
| Viga7/116 | F:(ROX-)GCCTGGTTGTGGATCTGTC R:ATCCTGCTACATCGACCTTC | 159–173 | 60 | |
| Vigt10/ε | F:(ROX-)GCAGTGCAGGAATAGTAAGG R:GCTGTGATACCAGAGAACGA | 171–173 | 60 | |
| Vict1/130 | F:(NED-)GATTGGTGACGCATGTGT R:GCTGGAGATTGCGTAGAC | 132–152 | 58 | |
| EMVi029 | F:(TAMRA-)ACGAGTCCCAGGTCTCACAG R:TGTTGACGGTCACGGTGTAT | 164–248 | 58 * | [32] |
| 1tc1g | F:(NED-)TCACTCAACAATACAGTTTCTTAG R:TTTCACGGTAGCGATAGGAG | 111–185 | 58 | [13] |
| 1tc1b | F:(HEX-)CGATTGGGGATATGAAGACTT R:TTAGTAATCAAATCGCACCCA | 149–210 | 58 | |
| 1tc1a | F:(FAM-)TCGAGATCCTCAAACTTCCTT R:TTTTAACTGTGCGGCCTG | 109–187 | 58 |
| Locus | Allele Size Range, bp | Na | Pa | Ne | I | h | PIC |
|---|---|---|---|---|---|---|---|
| Vicacg8/42 | 192–226 | 13 | 8 | 2.75 | 1.42 | 0.64 | 0.627 |
| Vitg11/70 | 186–210 | 11 | 6 | 5.79 | 1.96 | 0.83 | 0.823 |
| Vigtg10/95 | 128–157 | 9 | 4 | 2.30 | 1.14 | 0.57 | 0.549 |
| Vica9/152 | 152–185 | 9 | 4 | 2.65 | 1.42 | 0.62 | 0.602 |
| Vitc2D | 188–252 | 29 | 10 | 14.31 | 2.88 | 0.93 | 0.934 |
| Viga7/116 | 139–174 | 9 | 3 | 3.55 | 1.53 | 0.72 | 0.710 |
| Vigt10/ε | 174–176 | 2 | 1 | 1.01 | 0.04 | 0.01 | 0.013 |
| Vict1/130 | 149–157 | 5 | 1 | 1.68 | 0.87 | 0.40 | 0.405 |
| EMVi029 | 163–218 | 15 | 8 | 4.20 | 1.91 | 0.76 | 0.746 |
| 1tc1g | 112–188 | 27 | 15 | 9.27 | 2.63 | 0.89 | 0.884 |
| 1tc1b | 151–191 | 10 | 6 | 2.44 | 1.31 | 0.59 | 0.604 |
| 1tc1a | 109–160 | 19 | 8 | 13.62 | 2.73 | 0.93 | 0.925 |
| Group | N | Na | Pa | Ne | k | I | h |
|---|---|---|---|---|---|---|---|
| K1 | 54 | 7.33 | 1.67 | 3.60 | 14 | 1.33 | 0.59 |
| K2 | 40 | 6.08 | 0.92 | 3.71 | 11 | 1.18 | 0.53 |
| K3 | 49 | 9.00 | 2.75 | 4.57 | 14 | 1.51 | 0.64 |
| Source of Variation | Degrees of Freedom | Sum of Squares | Variance Components | Percentage Variation | PhiPT | Probability |
|---|---|---|---|---|---|---|
| Among populations (Zemgale and Kurzeme) | 2 | 17.33 | 0.21 | 5 | 0.052 | 0.001 |
| Within population | 141 | 546.74 | 3.88 | 95 | ||
| Total | 143 | 564.03 | 4.10 | 100 | ||
| Among populations (STRUCTURE K = 3) * | 3 | 57.41 | 0.53 | 13 | 0.128 | 0.001 |
| Within population | 140 | 507.65 | 3.63 | 87 | ||
| Total | 143 | 565.06 | 4.16 | 100 | ||
| Among populations (STRUCTURE K = 4) ** | 4 | 76.10 | 0.61 | 15 | 0.147 | 0.001 |
| Within Pops | 145 | 511.69 | 3.53 | 85 | ||
| Total | 149 | 587.79 | 4.14 | 100 |
| Group (K) | Parameter | Vicacg8/42 | Vitg11/70 | Vigtg10/95 | Vica9/152 | Vitc2/D | Viga7/116 | Vigt10/ε | Vict1/130 | EMVi029 | 1tc1g | 1tc1b | 1tc1a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K1 | N | 54 | 54 | 54 | 54 | 54 | 54 | 54 | 54 | 54 | 54 | 54 | 54 |
| Na | 5 | 5 | 6 | 7 | 13 | 6 | 1 | 4 | 8 | 13 | 7 | 13 | |
| Ne | 1.42 | 2.62 | 2.12 | 3.65 | 8.84 | 3.88 | 1.00 | 1.42 | 3.70 | 3.58 | 3.04 | 7.92 | |
| I | 0.65 | 1.18 | 1.05 | 1.53 | 2.33 | 1.50 | 0.00 | 0.63 | 1.65 | 1.85 | 1.35 | 2.25 | |
| h | 0.30 | 0.62 | 0.53 | 0.73 | 0.89 | 0.74 | 0.00 | 0.30 | 0.73 | 0.72 | 0.67 | 0.87 | |
| K2 | N | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 40 |
| Na | 5 | 6 | 4 | 5 | 13 | 3 | 1 | 2 | 8 | 13 | 3 | 10 | |
| Ne | 1.71 | 3.32 | 2.43 | 1.53 | 8.51 | 2.89 | 1.00 | 1.05 | 5.63 | 7.69 | 1.23 | 7.55 | |
| I | 0.85 | 1.43 | 1.01 | 0.74 | 2.32 | 1.08 | 0.00 | 0.12 | 1.84 | 2.25 | 0.38 | 2.14 | |
| h | 0,42 | 0.70 | 0.59 | 0.35 | 0.88 | 0.65 | 0.00 | 0.05 | 0.82 | 0.87 | 0.18 | 0.87 | |
| K3 | N | 49 | 49 | 49 | 49 | 49 | 49 | 49 | 49 | 49 | 49 | 49 | 49 |
| Na | 9 | 9 | 6 | 7 | 19 | 5 | 2 | 5 | 6 | 20 | 6 | 14 | |
| Ne | 2.65 | 4.95 | 2.04 | 2.72 | 12.84 | 2.27 | 1.04 | 2.99 | 2.26 | 9.49 | 2.57 | 9.06 | |
| I | 1.46 | 1.81 | 0.99 | 1.38 | 2.72 | 0.99 | 0.10 | 1.31 | 1.09 | 2.60 | 1.24 | 2.39 | |
| h | 0.62 | 0.80 | 0.51 | 0.63 | 0.92 | 0.56 | 0.04 | 0.67 | 0.56 | 0.89 | 0.61 | 0.89 |
| Group (K) | N | Na | Pa | Ne | k | I | h |
|---|---|---|---|---|---|---|---|
| K1 | 23 | 4.67 | 0.42 | 2.94 | 7 | 1.03 | 0.50 |
| K2 | 36 | 8.00 | 2.00 | 4.13 | 14 | 1.42 | 0.62 |
| K3 | 58 | 7.67 | 1.83 | 3.92 | 14 | 1.32 | 0.58 |
| K4 | 32 | 5.42 | 0.75 | 3.10 | 9 | 1.17 | 0.56 |
| Group (K) | Parameter | Vicacg8/42 | Vitg11/70 | Vigtg10/95 | Vica9/152 | Vitc2/D | Viga7/116 | Vigt10/ε | Vict1/130 | EMVi029 | 1tc1g | 1tc1b | 1tc1a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K1 | N | 23 | 23 | 23 | 23 | 23 | 23 | 23 | 23 | 23 | 23 | 23 | 23 |
| Na | 4 | 5 | 3 | 3 | 9 | 4 | 1 | 2 | 8 | 8 | 3 | 6 | |
| Ne | 1.44 | 3.33 | 1.71 | 1.43 | 7.05 | 2.33 | 1.00 | 1.19 | 5.45 | 4.52 | 1.43 | 4.45 | |
| I | 0.64 | 1.35 | 0.74 | 0.56 | 2.05 | 0.98 | 0.00 | 0.30 | 1.84 | 1.74 | 0.56 | 1.62 | |
| h | 0.31 | 0.70 | 0.42 | 0.30 | 0.86 | 0.57 | 0.00 | 0.16 | 0.82 | 0.78 | 0.30 | 0.78 | |
| K2 | N | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 |
| Na | 10 | 9 | 3 | 6 | 16 | 4 | 1 | 4 | 7 | 17 | 6 | 13 | |
| Ne | 2.75 | 3.70 | 1.66 | 2.27 | 9.00 | 2.20 | 1.00 | 2.81 | 2.72 | 11.17 | 2.87 | 7.36 | |
| I | 1.55 | 1.70 | 0.69 | 1.20 | 2.47 | 0.90 | 0.00 | 1.20 | 1.27 | 2.61 | 1.26 | 2.25 | |
| h | 0.64 | 0.73 | 0.40 | 0.56 | 0.89 | 0.55 | 0.00 | 0.64 | 0.63 | 0.91 | 0.65 | 0.86 | |
| K3 | N | 58 | 58 | 58 | 58 | 58 | 58 | 58 | 58 | 58 | 58 | 58 | 58 |
| Na | 6 | 9 | 4 | 6 | 19 | 3 | 2 | 3 | 8 | 15 | 5 | 12 | |
| Ne | 2.96 | 4.38 | 1.57 | 2.25 | 10.32 | 2.15 | 1.04 | 1.41 | 2.91 | 8.09 | 1.72 | 8.25 | |
| I | 1.30 | 1.72 | 0.65 | 1.15 | 2.64 | 0.85 | 0.09 | 0.52 | 1.43 | 2.35 | 0.83 | 2.27 | |
| h | 0.66 | 0.77 | 0.36 | 0.56 | 0.90 | 0.54 | 0.03 | 0.29 | 0.66 | 0.88 | 0.42 | 0.88 | |
| K4 | N | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 |
| Na | 3 | 5 | 5 | 6 | 10 | 5 | 1 | 4 | 5 | 7 | 4 | 10 | |
| Ne | 1.14 | 2.28 | 2.81 | 3.58 | 7.21 | 3.35 | 1.00 | 1.71 | 3.68 | 1.88 | 2.99 | 5.63 | |
| I | 0.28 | 1.11 | 1.21 | 1.45 | 2.12 | 1.39 | 0.00 | 0.83 | 1.41 | 1.07 | 1.20 | 2.00 | |
| h | 0.12 | 0.56 | 0.64 | 0.72 | 0.86 | 0.70 | 0.00 | 0.42 | 0.73 | 0.47 | 0.67 | 0.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokolova, O.; Moročko-Bičevska, I.; Lācis, G. Genetic Diversity of Venturia inaequalis in Latvia Revealed by Microsatellite Markers. Pathogens 2022, 11, 1165. https://doi.org/10.3390/pathogens11101165
Sokolova O, Moročko-Bičevska I, Lācis G. Genetic Diversity of Venturia inaequalis in Latvia Revealed by Microsatellite Markers. Pathogens. 2022; 11(10):1165. https://doi.org/10.3390/pathogens11101165
Chicago/Turabian StyleSokolova, Olga, Inga Moročko-Bičevska, and Gunārs Lācis. 2022. "Genetic Diversity of Venturia inaequalis in Latvia Revealed by Microsatellite Markers" Pathogens 11, no. 10: 1165. https://doi.org/10.3390/pathogens11101165
APA StyleSokolova, O., Moročko-Bičevska, I., & Lācis, G. (2022). Genetic Diversity of Venturia inaequalis in Latvia Revealed by Microsatellite Markers. Pathogens, 11(10), 1165. https://doi.org/10.3390/pathogens11101165