
Alphavirus Virulence Determinants

Abstract
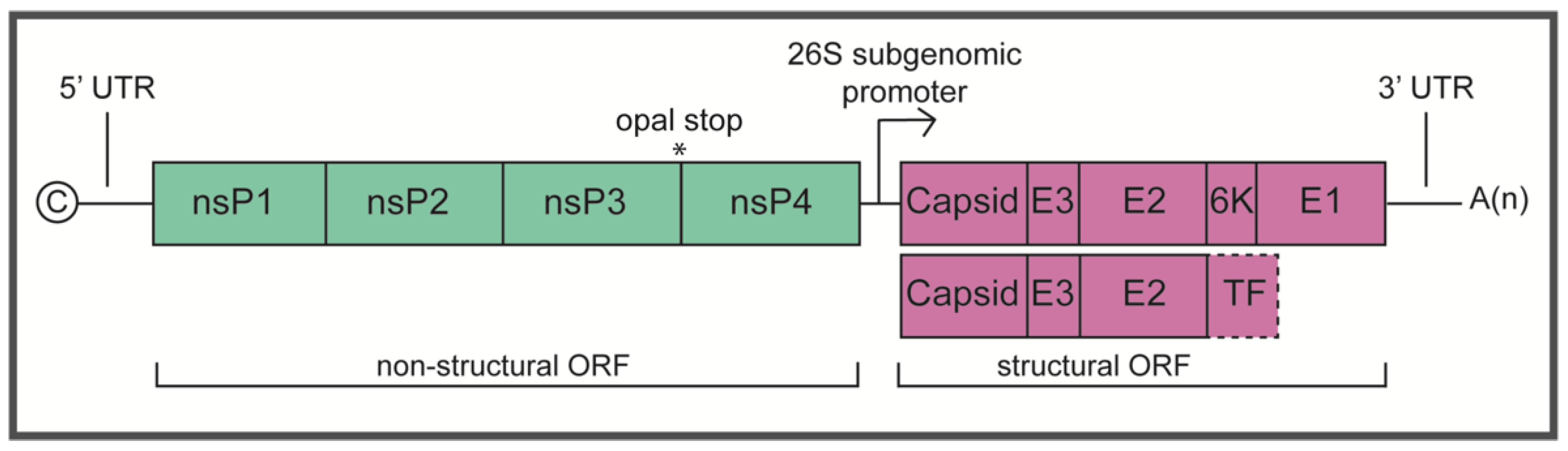
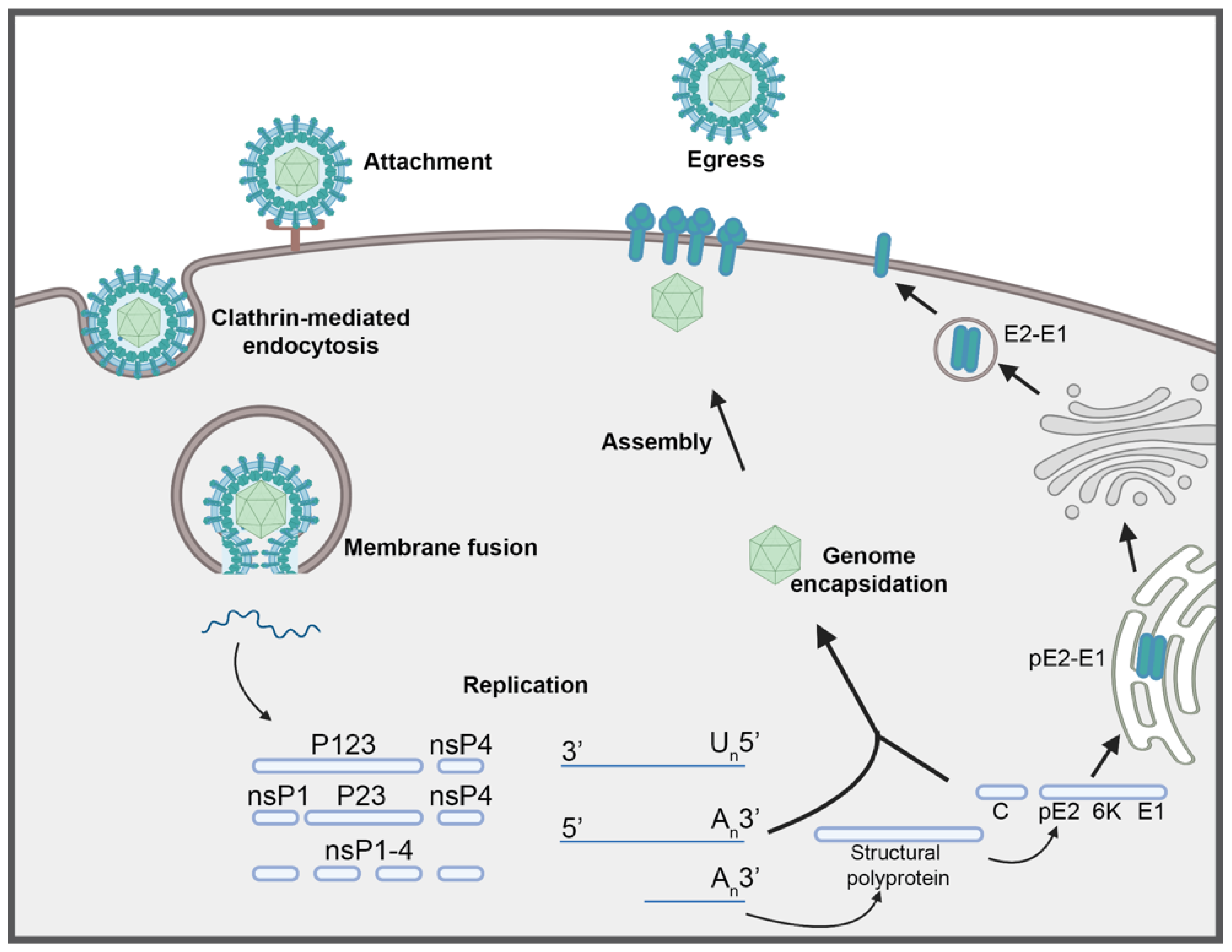
:1. Introduction
2. Determinants of Virulence
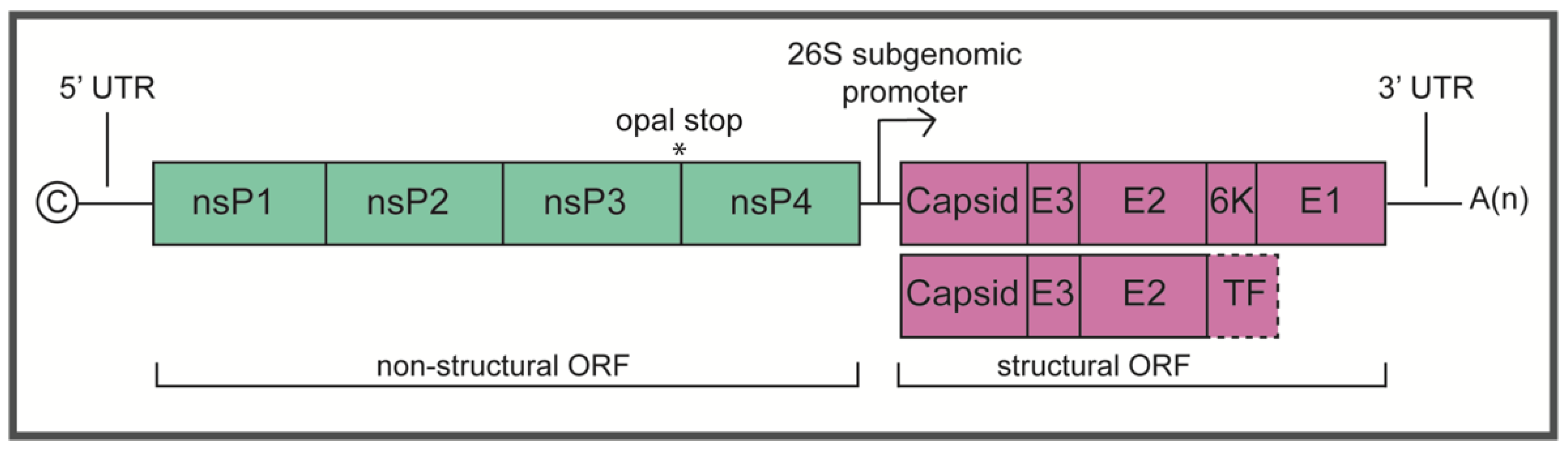
2.1. Non-Structural Proteins
2.1.1. nsP1
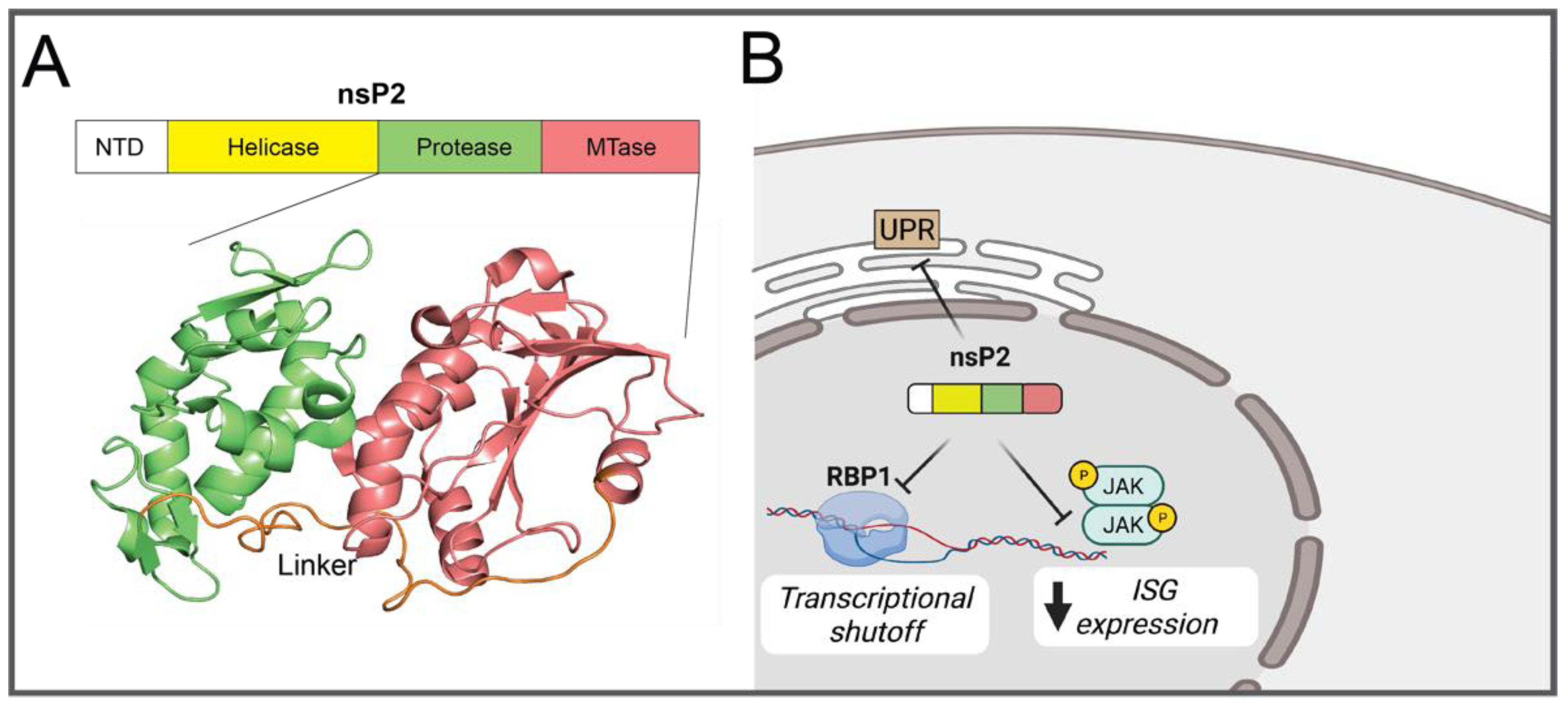
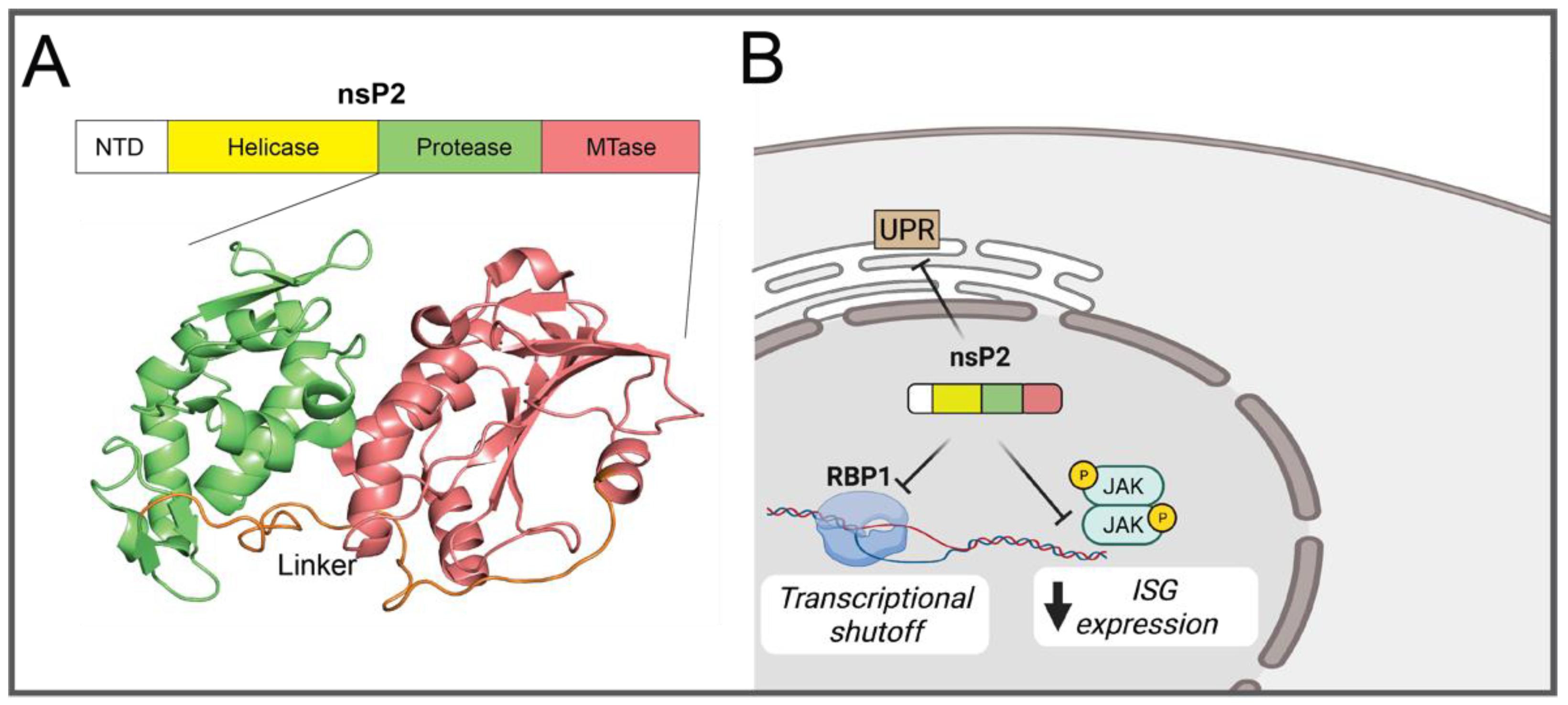
2.1.2. nsP2
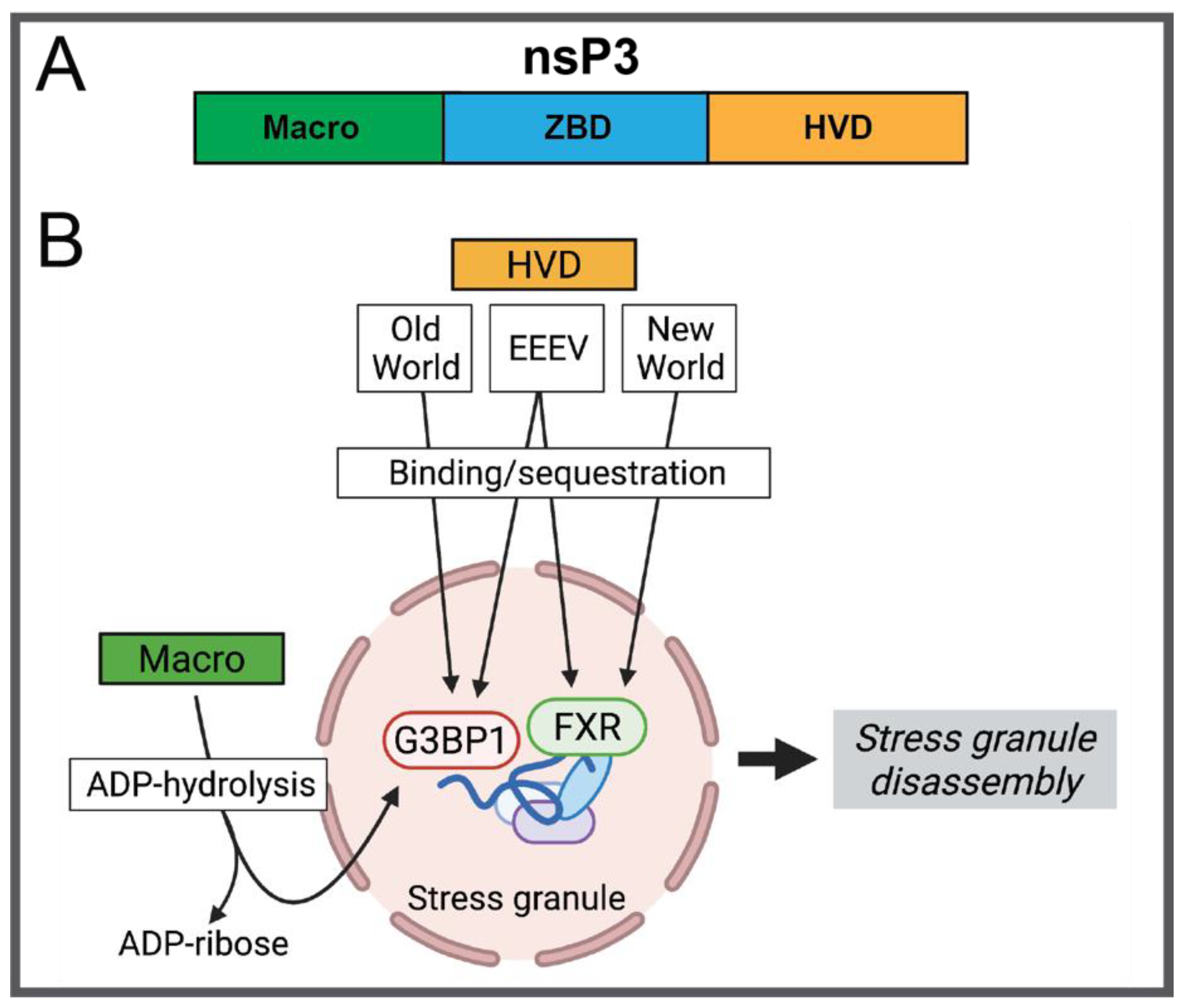
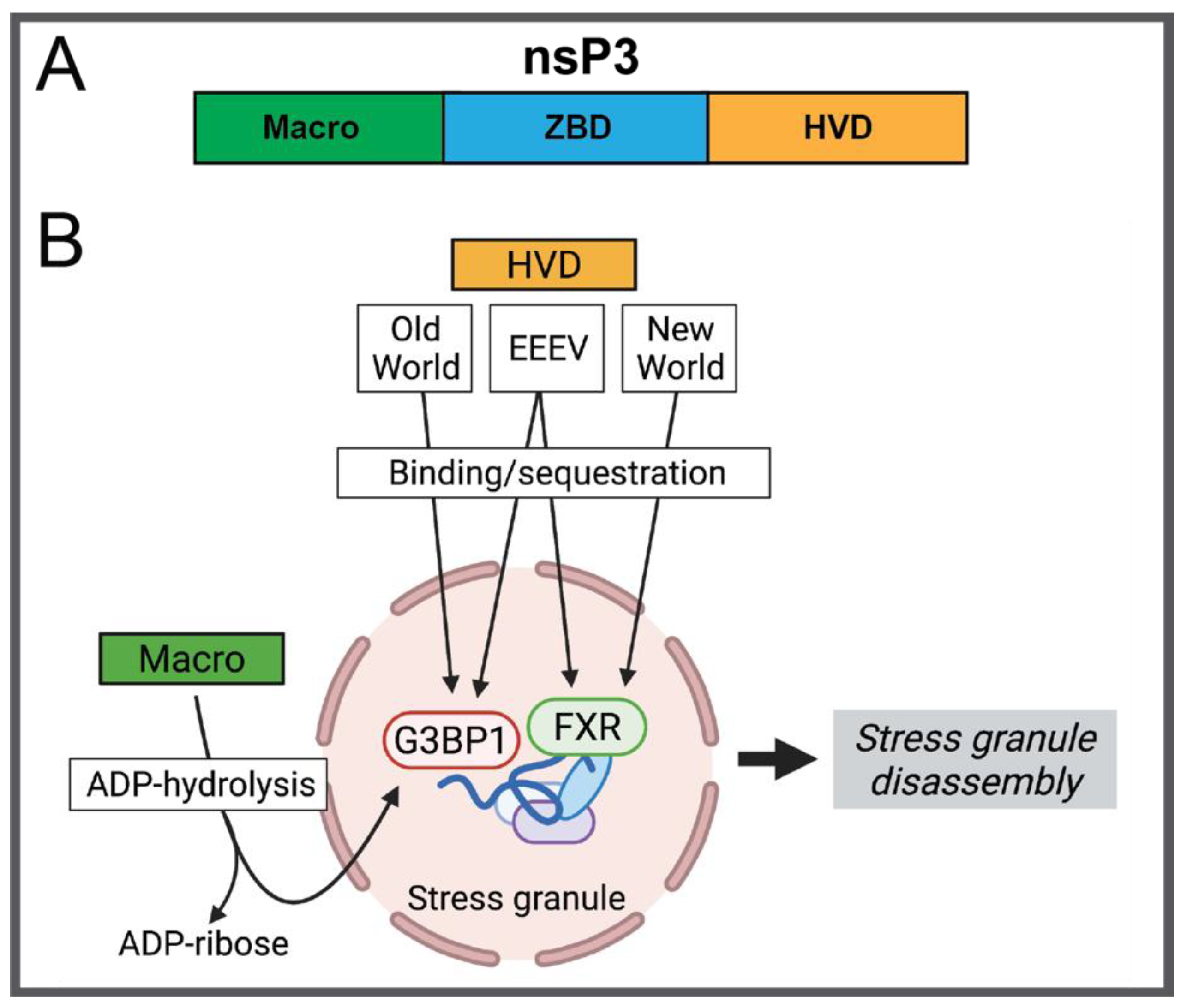
2.1.3. nsP3
2.1.4. nsP4
2.2. Structural Proteins
2.2.1. Capsid
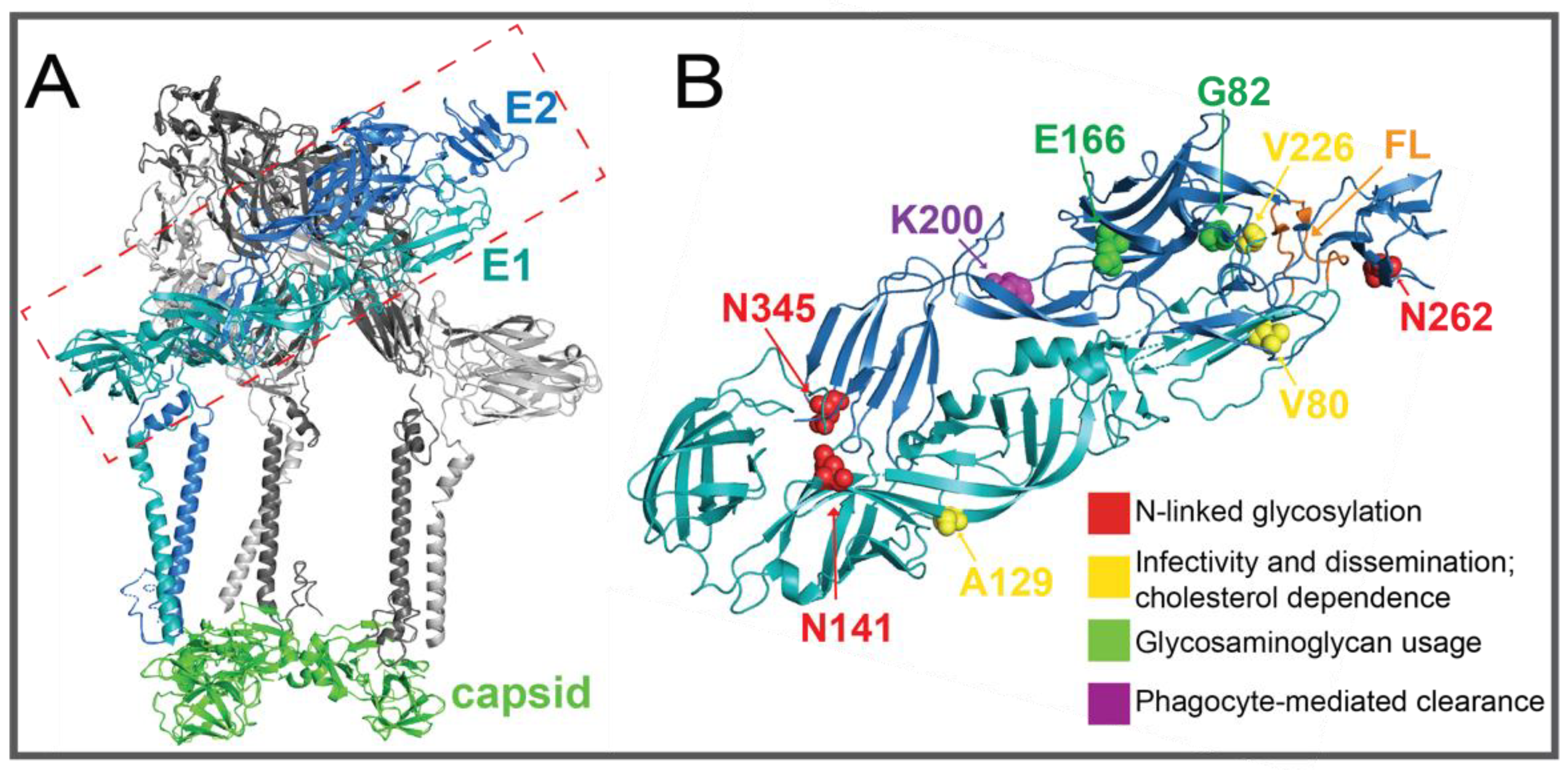
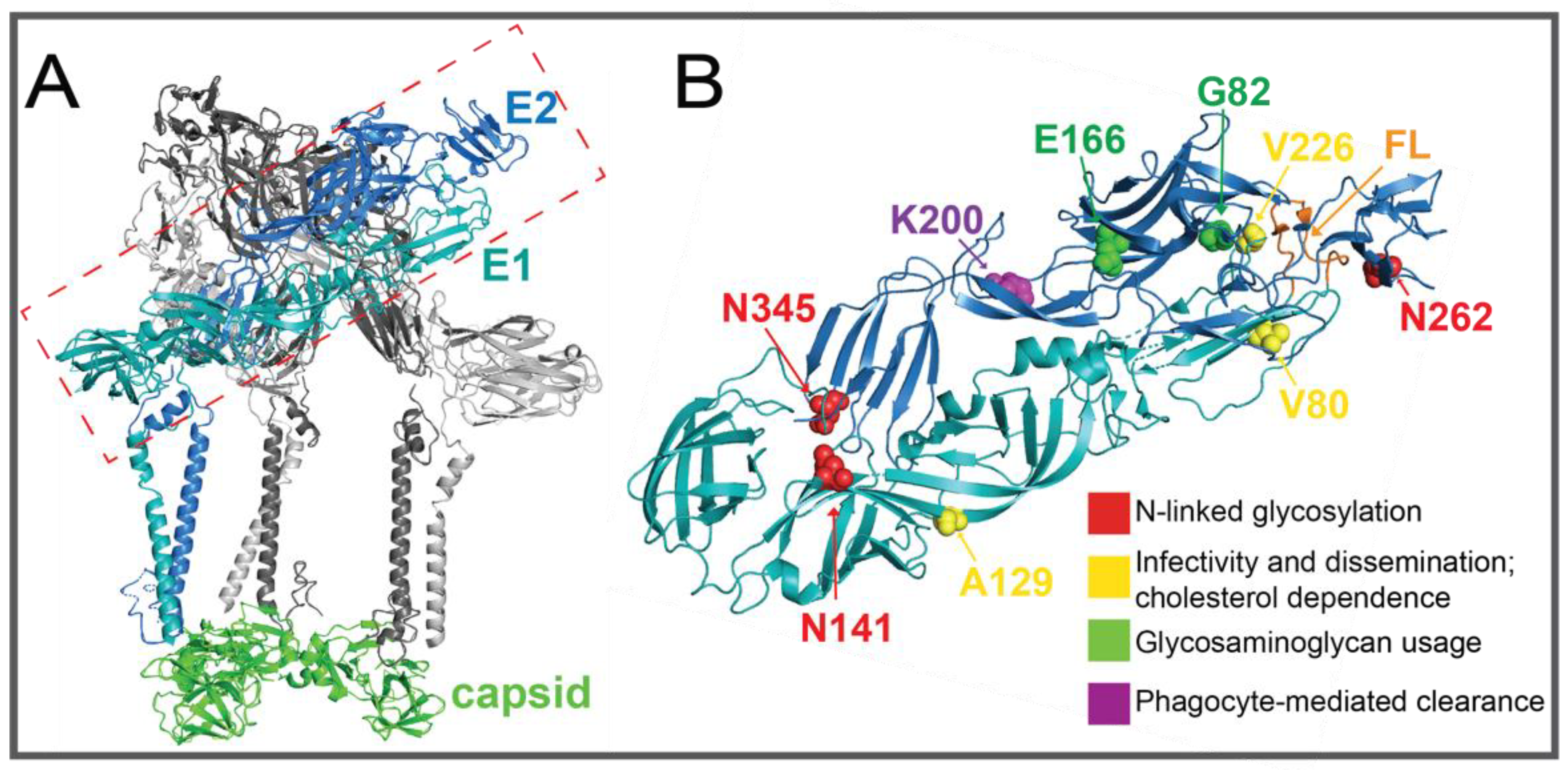
2.2.2. E1 and E2 Glycoproteins
2.2.3. E3
2.2.4. 6K and TF
2.3. 5′ and 3′ Untranslated Regions
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sudeep, A.B. Culex gelidus: An emerging mosquito vector with potential to transmit multiple virus infections. J. Vector Borne Dis. 2014, 51, 251–258. [Google Scholar]
- Johnson, B.K.; Gichogo, A.; Gitau, G.; Patel, N.; Ademba, G.; Kirui, R.; Highton, R.B.; Smith, D.H. Recovery of o’nyong-nyong virus from Anopheles funestus in Western Kenya. Trans. R Soc. Trop. Med. Hyg. 1981, 75, 239–241. [Google Scholar] [CrossRef]
- Lounibos, L.P.; Kramer, L.D. Invasiveness of Aedes aegypti and Aedes albopictus and Vectorial Capacity for Chikungunya Virus. J. Infect Dis. 2016, 214, S453–S458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, M.U.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef] [PubMed]
- Tatem, A.J.; Hay, S.I.; Rogers, D.J. Global traffic and disease vector dispersal. Proc. Natl. Acad. Sci. USA 2006, 103, 6242–6247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkett-Cadena, N.D.; Vittor, A.Y. Deforestation and vector-borne disease: Forest conversion favors important mosquito vectors of human pathogens. Basic Appl. Ecol. 2018, 26, 101–110. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Weaver, S.C. Sequential adaptive mutations enhance efficient vector switching by Chikungunya virus and its epidemic emergence. PLoS Pathog. 2011, 7, e1002412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanciotti, R.S.; Valadere, A.M. Transcontinental movement of Asian genotype chikungunya virus. Emerg. Infect. Dis. 2014, 20, 1400. [Google Scholar] [CrossRef]
- Levi, L.I.; Vignuzzi, M. Arthritogenic Alphaviruses: A Worldwide Emerging Threat? Microorganisms 2019, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Zacks, M.A.; Paessler, S. Encephalitic alphaviruses. Vet. Microbiol. 2010, 140, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Langsjoen, R.M.; Haller, S.L.; Roy, C.J.; Vinet-Oliphant, H.; Bergren, N.A.; Erasmus, J.H.; Livengood, J.A.; Powell, T.D.; Weaver, S.C.; Rossi, S.L. Chikungunya Virus Strains Show Lineage-Specific Variations in Virulence and Cross-Protective Ability in Murine and Nonhuman Primate Models. mBio 2018, 9, e02449-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.C.; Basore, K.; Fremont, D.H.; Diamond, M.S. A molecular understanding of alphavirus entry. PLoS Pathog. 2020, 16, e1008876. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Solignat, M.; Gay, B.; Chazal, N.; Higgs, S.; Devaux, C.; Briant, L. Endocytosis of chikungunya virus into mammalian cells: Role of clathrin and early endosomal compartments. PLoS ONE 2010, 5, e11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielian, M.; Chanel-Vos, C.; Liao, M. Alphavirus entry and membrane fusion. Viruses 2010, 2, 796–825. [Google Scholar] [CrossRef] [Green Version]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 1988, 107, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spuul, P.; Balistreri, G.; Kääriäinen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef] [Green Version]
- Pietilä, M.K.; Hellström, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, J.; Mukhopadhyay, S. Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins. Viruses 2017, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Melancon, P.; Garoff, H. Processing of the Semliki Forest virus structural polyprotein: Role of the capsid protease. J. Virol. 1987, 61, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, M.; Brown, D.T. Assembly of the Sindbis virus spike protein complex. Virology 1996, 219, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S.; Wan, J.J.; Kielian, M. The Alphavirus Exit Pathway: What We Know and What We Wish We Knew. Viruses 2018, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Tuittila, M.T.; Santagati, M.G.; Röyttä, M.; Määttä, J.A.; Hinkkanen, A.E. Replicase complex genes of Semliki Forest virus confer lethal neurovirulence. J. Virol. 2000, 74, 4579–4589. [Google Scholar] [CrossRef]
- Atkins, G.J.; Sheahan, B.J. Molecular determinants of alphavirus neuropathogenesis in mice. J. Gen. Virol. 2016, 97, 1283–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heise, M.T.; Simpson, D.A.; Johnston, R.E. A single amino acid change in nsP1 attenuates neurovirulence of the Sindbis-group alphavirus S.A.AR86. J. Virol. 2000, 74, 4207–4213. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.C.; Suthar, M.S.; Montgomery, S.A.; Shabman, R.; Simmons, J.; Johnston, R.E.; Morrison, T.E.; Heise, M.T. Modulation of type I IFN induction by a virulence determinant within the alphavirus nsP1 protein. Virology 2010, 399, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoermer Burrack, K.A.; Hawman, D.W.; Jupille, H.J.; Oko, L.; Minor, M.; Shives, K.D.; Gunn, B.M.; Long, K.M.; Morrison, T.E. Attenuating Mutations in nsP1 Reveal Tissue-Specific Mechanisms for Control of Ross River Virus Infection. J. Virol. 2014, 88, 3719–3732. [Google Scholar] [CrossRef] [Green Version]
- Frolova, E.I.; Fayzulin, R.Z.; Cook, S.H.; Griffin, D.E.; Rice, C.M.; Frolov, I. Roles of nonstructural protein nsP2 and Alpha/Beta interferons in determining the outcome of Sindbis virus infection. J. Virol. 2002, 76, 11254–11264. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.Y.; Fu, S.H.; Wang, J.L.; He, Y.; Tang, Q.; Liang, G.D. Effects of the nsP2-726 Pro mutation on infectivity and pathogenesis of Sindbis virus derived from a full-length infectious cDNA clone. Virus Res. 2009, 142, 204–207. [Google Scholar] [CrossRef]
- Fros, J.J.; Liu, W.J.; Prow, N.A.; Geertsema, C.; Ligtenberg, M.; Vanlandingham, D.L.; Schnettler, E.; Vlak, J.M.; Suhrbier, A.; Khromykh, A.A.; et al. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J. Virol. 2010, 84, 10877–10887. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Mutso, M.; Cherkashchenko, L.; Zusinaite, E.; Herrero, L.J.; Doggett, S.L.; Haniotis, J.; Merits, A.; Herring, B.L.; Taylor, A.; et al. Identification of Natural Molecular Determinants of Ross River Virus Type I Interferon Modulation. J. Virol. 2020, 94, e01788-19. [Google Scholar] [CrossRef]
- Akhrymuk, I.; Lukash, T.; Frolov, I.; Frolova, E.I. Novel Mutations in nsP2 Abolish Chikungunya Virus-Induced Transcriptional Shutoff and Make the Virus Less Cytopathic without Affecting Its Replication Rates. J. Virol. 2019, 93, e02062-18. [Google Scholar] [CrossRef] [Green Version]
- Fros, J.J.; Major, L.D.; Scholte, F.E.M.; Gardner, J.; van Hemert, M.J.; Suhrbier, A.; Pijlman, G.P. Chikungunya virus non-structural protein 2-mediated host shut-off disables the unfolded protein response. J. Gen. Virol. 2015, 96, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihinen, H.; Ahola, T.; Tuittila, M.; Merits, A.; Kääriäinen, L. Elimination of phosphorylation sites of Semliki Forest virus replicase protein nsP3. J. Biol. Chem. 2001, 276, 5745–5752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suthar, M.S.; Shabman, R.; Madric, K.; Lambeth, C.; Heise, M.T. Identification of adult mouse neurovirulence determinants of the Sindbis virus strain AR86. J. Virol. 2005, 79, 4219–4228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuittila, M.; Hinkkanen, A.E. Amino acid mutations in the replicase protein nsP3 of Semliki Forest virus cumulatively affect neurovirulence. J. Gen. Virol. 2003, 84, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.E.; Long, K.M.; Whitmore, A.C.; Sanders, W.; Thurlow, L.R.; Brown, J.A.; Morrison, C.R.; Vincent, H.; Peck, K.M.; Browning, C.; et al. Disruption of the Opal Stop Codon Attenuates Chikungunya Virus-Induced Arthritis and Pathology. mBio 2017, 8, e01456-17. [Google Scholar] [CrossRef] [Green Version]
- Abraham, R.; McPherson, R.L.; Dasovich, M.; Badiee, M.; Leung, A.K.L.; Griffin, D.E. Both ADP-Ribosyl-Binding and Hydrolase Activities of the Alphavirus nsP3 Macrodomain Affect Neurovirulence in Mice. mBio 2020, 11, e03253-19. [Google Scholar] [CrossRef] [Green Version]
- Meshram, C.D.; Shiliaev, N.; Frolova, E.I.; Frolov, I. Hypervariable Domain of nsP3 of Eastern Equine Encephalitis Virus Is a Critical Determinant of Viral Virulence. J. Virol. 2020, 94, e00617-20. [Google Scholar] [CrossRef]
- Coffey, L.L.; Beeharry, Y.; Bordería, A.V.; Blanc, H.; Vignuzzi, M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16038–16043. [Google Scholar] [CrossRef] [Green Version]
- Warmbrod, K.L.; Patterson, E.I.; Kautz, T.F.; Stanton, A.; Rockx-Brouwer, D.; Kalveram, B.K.; Khanipov, K.; Thangamani, S.; Fofanov, Y.; Forrester, N.L. Viral RNA-dependent RNA polymerase mutants display an altered mutation spectrum resulting in attenuation in both mosquito and vertebrate hosts. PLoS Pathog. 2019, 15, e1007610. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.P.S.; Ng, M.-L.; Vasudevan, S.G. Differential unfolded protein response during Chikungunya and Sindbis virus infection: CHIKV nsP4 suppresses eIF2α phosphorylation. Virol. J. 2013, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Atasheva, S.; Garmashova, N.; Frolov, I.; Frolova, E. Venezuelan equine encephalitis virus capsid protein inhibits nuclear import in Mammalian but not in mosquito cells. J. Virol. 2008, 82, 4028–4041. [Google Scholar] [CrossRef] [Green Version]
- Atasheva, S.; Fish, A.; Fornerod, M.; Frolova, E.I. Venezuelan equine Encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. J. Virol. 2010, 84, 4158–4171. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, P.V.; Leung, L.W.; Wang, E.; Weaver, S.C.; Basler, C.F. A five-amino-acid deletion of the eastern equine encephalitis virus capsid protein attenuates replication in mammalian systems but not in mosquito cells. J. Virol. 2008, 82, 6972–6983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltier, D.C.; Lazear, H.M.; Farmer, J.R.; Diamond, M.S.; Miller, D.J. Neurotropic arboviruses induce interferon regulatory factor 3-mediated neuronal responses that are cytoprotective, interferon independent, and inhibited by Western equine encephalitis virus capsid. J. Virol. 2013, 87, 1821–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.B.; Weaver, S.C. Structure of the recombinant alphavirus Western equine encephalitis virus revealed by cryoelectron microscopy. J. Virol. 2010, 84, 9775–9782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Q.; Zhou, H.; Shu, T.; Mu, J.; Fang, Y.; Xu, J.; Li, T.; Kong, J.; Qiu, Y.; Zhou, X. The Capsid Protein of Semliki Forest Virus Antagonizes RNA Interference in Mammalian Cells. J. Virol. 2020, 94, e01233-19. [Google Scholar] [CrossRef] [Green Version]
- Lustig, S.; Jackson, A.C.; Hahn, C.S.; Griffin, D.E.; Strauss, E.G.; Strauss, J.H. Molecular basis of Sindbis virus neurovirulence in mice. J. Virol. 1988, 62, 2329–2336. [Google Scholar] [CrossRef] [Green Version]
- Stapleford, K.A.; Coffey, L.L.; Lay, S.; Bordería, A.V.; Duong, V.; Isakov, O.; Rozen-Gagnon, K.; Arias-Goeta, C.; Blanc, H.; Beaucourt, S. Emergence and transmission of arbovirus evolutionary intermediates with epidemic potential. Cell Host Microbe 2014, 15, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Noval, M.G.; Rodriguez-Rodriguez, B.A.; Rangel, M.V.; Stapleford, K.A. Evolution-Driven Attenuation of Alphaviruses Highlights Key Glycoprotein Determinants Regulating Viral Infectivity and Dissemination. Cell Rep. 2019, 28, 460–471.e465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; McGee, C.E.; Higgs, S. Chikungunya virus adaptation to Aedes albopictus mosquitoes does not correlate with acquisition of cholesterol dependence or decreased pH threshold for fusion reaction. Virol. J. 2011, 8, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashishtha, M.; Phalen, T.; Marquardt, M.T.; Ryu, J.S.; Ng, A.C.; Kielian, M. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J. Cell Biol. 1998, 140, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.E.; Cassese, T.; Kielian, M. The cholesterol requirement for sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 1999, 73, 4272–4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.A.; Herrero, L.J.; Jeffery, J.A.L.; Hoehn, M.; Rudd, P.A.; Supramaniam, A.; Kay, B.H.; Ryan, P.A.; Mahalingam, S. Role of envelope N-linked glycosylation in Ross River virus virulence and transmission. J. Gen. Virol. 2016, 97, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Ashbrook, A.W.; Burrack, K.S.; Silva, L.A.; Montgomery, S.A.; Heise, M.T.; Morrison, T.E.; Dermody, T.S. Residue 82 of the Chikungunya virus E2 attachment protein modulates viral dissemination and arthritis in mice. J. Virol. 2014, 88, 12180–12192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, L.A.; Khomandiak, S.; Ashbrook, A.W.; Weller, R.; Heise, M.T.; Morrison, T.E.; Dermody, T.S. A single-amino-acid polymorphism in Chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J. Virol. 2014, 88, 2385–2397. [Google Scholar] [CrossRef] [Green Version]
- Gad, H.H.; Paulous, S.; Belarbi, E.; Diancourt, L.; Drosten, C.; Kümmerer, B.M.; Plate, A.E.; Caro, V.; Desprès, P. The E2-E166K substitution restores Chikungunya virus growth in OAS3 expressing cells by acting on viral entry. Virology 2012, 434, 27–37. [Google Scholar]
- Gardner, C.L.; Ebel, G.D.; Ryman, K.D.; Klimstra, W.B. Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc. Natl. Acad. Sci. USA 2011, 108, 16026–16031. [Google Scholar] [CrossRef] [Green Version]
- Hawman, D.W.; Carpentier, K.S.; Fox, J.M.; May, N.A.; Sanders, W.; Montgomery, S.A.; Moorman, N.J.; Diamond, M.S.; Morrison, T.E. Mutations in the E2 Glycoprotein and the 3′ Untranslated Region Enhance Chikungunya Virus Virulence in Mice. J. Virol. 2017, 91, e00816-17. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, K.S.; Davenport, B.J.; Haist, K.C.; McCarthy, M.K.; May, N.A.; Robison, A.; Ruckert, C.; Ebel, G.D.; Morrison, T.E. Discrete viral E2 lysine residues and scavenger receptor MARCO are required for clearance of circulating alphaviruses. eLife 2019, 8, e49163. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.L.; Schultz, K.L.W.; Kent, R.J.; Venkatesan, M.; Griffin, D.E. Role of N-linked glycosylation for sindbis virus infection and replication in vertebrate and invertebrate systems. J. Virol. 2009, 83, 5640–5647. [Google Scholar] [CrossRef] [Green Version]
- Heidner, H.W.; McKnight, K.L.; Davis, N.L.; Johnston, R.E. Lethality of PE2 incorporation into Sindbis virus can be suppressed by second-site mutations in E3 and E2. J. Virol. 1994, 68, 2683–2692. [Google Scholar] [CrossRef] [Green Version]
- Hallengärd, D.; Kakoulidou, M.; Lulla, A.; Kümmerer, B.M.; Johansson, D.X.; Mutso, M.; Lulla, V.; Fazakerley, J.K.; Roques, P.; Le Grand, R.; et al. Novel attenuated Chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. J. Virol. 2014, 88, 2858–2866. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Melton, J.V.; Herrero, L.J.; Thaa, B.; Karo-Astover, L.; Gage, P.W.; Nelson, M.A.; Sheng, K.-C.; Lidbury, B.A.; Ewart, G.D.; et al. Effects of an In-Frame Deletion of the 6k Gene Locus from the Genome of Ross River Virus. J. Virol. 2016, 90, 4150–4159. [Google Scholar] [CrossRef] [Green Version]
- Kendra, J.A.; de la Fuente, C.; Brahms, A.; Woodson, C.; Bell, T.M.; Chen, B.; Khan, Y.A.; Jacobs, J.L.; Kehn-Hall, K.; Dinman, J.D. Ablation of Programmed -1 Ribosomal Frameshifting in Venezuelan Equine Encephalitis Virus Results in Attenuated Neuropathogenicity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.J.; Jones-Burrage, S.; Maury, W.; Mukhopadhyay, S. TF protein of Sindbis virus antagonizes host type I interferon responses in a palmitoylation-dependent manner. Virology 2020, 542, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.; Chavez, M.; Mukhopadhyay, S. Domains of the TF protein important in regulating its own palmitoylation. Virology 2019, 531, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, D.; Rice, C.M.; Brodie, C.; Shahar, A.; Dubuisson, J.; Halevy, M.; Lustig, S. A single nucleotide change in the 5′ noncoding region of Sindbis virus confers neurovirulence in rats. J. Virol. 1999, 73, 10440–10446. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, R.J.; Griffin, D.E.; Zhang, H.; Niesters, H.G.; Strauss, J.H. Attenuation of Sindbis virus neurovirulence by using defined mutations in nontranslated regions of the genome RNA. J. Virol. 1992, 66, 7121–7127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, C.H.; Sheahan, B.J.; Atkins, G.J. The 5′ untranslated region as a pathogenicity determinant of Semliki Forest virus in mice. Virus Genes 2008, 36, 313–321. [Google Scholar] [CrossRef]
- White, L.J.; Wang, J.-G.; Davis, N.L.; Johnston, R.E. Role of Alpha/Beta Interferon in Venezuelan Equine Encephalitis Virus Pathogenesis: Effect of an Attenuating Mutation in the 5′ Untranslated Region. J. Virol. 2001, 75, 3706–3718. [Google Scholar] [CrossRef] [Green Version]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.W.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trobaugh, D.W.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, R.J.; Hong, Z.; Strauss, J.H. Mutagenesis of the 3′ nontranslated region of Sindbis virus RNA. J. Virol. 1990, 64, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Guillén, J.; Rabah, N.; Blanjoie, A.; Debart, F.; Vasseur, J.-J.; Canard, B.; Decroly, E.; Coutard, B. mRNA Capping by Venezuelan Equine Encephalitis Virus nsP1: Functional Characterization and Implications for Antiviral Research. J. Virol. 2015, 89, 8292–8303. [Google Scholar] [CrossRef] [Green Version]
- Jupille, H.J.; Oko, L.; Stoermer, K.A.; Heise, M.T.; Mahalingam, S.; Gunn, B.M.; Morrison, T.E. Mutations in nsP1 and PE2 are critical determinants of Ross River virus-induced musculoskeletal inflammatory disease in a mouse model. Virology 2011, 410, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, L.G.; Veloz, J.; Pintado-Silva, J.; Zhu, T.; Rangel, M.V.; Mutetwa, T.; Zhang, L.; Bernal-Rubio, D.; Figueroa, D.; Carrau, L.; et al. Chikungunya virus antagonizes cGAS-STING mediated type-I interferon responses by degrading cGAS. PLoS Pathog. 2020, 16, e1008999. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Ahola, T.; Kääriäinen, L. The effects of palmitoylation on membrane association of Semliki forest virus RNA capping enzyme. J. Biol. Chem. 1996, 271, 28567–28571. [Google Scholar] [CrossRef] [Green Version]
- Žusinaite, E.; Tints, K.; Kiiver, K.; Spuul, P.; Karo-Astover, L.; Merits, A.; Sarand, I. Mutations at the palmitoylation site of non-structural protein nsP1 of Semliki Forest virus attenuate virus replication and cause accumulation of compensatory mutations. J. Gen. Virol. 2007, 88, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.; Ferguson, M.; Cordonin, C.; Fragkoudis, R.; Ool, M.; Tamberg, N.; Sherwood, K.; Fazakerley, J.K.; Merits, A. Differences in Processing Determinants of Nonstructural Polyprotein and in the Sequence of Nonstructural Protein 3 Affect Neurovirulence of Semliki Forest Virus. J. Virol. 2015, 89, 11030–11045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rausalu, K.; Utt, A.; Quirin, T.; Varghese, F.S.; Žusinaite, E.; Das, P.K.; Ahola, T.; Merits, A. Chikungunya virus infectivity, RNA replication and non-structural polyprotein processing depend on the nsP2 protease’s active site cysteine residue. Sci. Rep. 2016, 6, 37124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez de Cedrón, M.; Ehsani, N.; Mikkola, M.L.; García, J.A.; Kääriäinen, L. RNA helicase activity of Semliki Forest virus replicase protein NSP2. FEBS Lett. 1999, 448, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Vasiljeva, L.; Merits, A.; Auvinen, P.; Kääriäinen, L. Identification of a novel function of the alphavirus capping apparatus. RNA 5′-triphosphatase activity of Nsp2. J. Biol. Chem. 2000, 275, 17281–17287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narwal, M.; Singh, H.; Pratap, S.; Malik, A.; Kuhn, R.J.; Kumar, P.; Tomar, S. Crystal structure of chikungunya virus nsP2 cysteine protease reveals a putative flexible loop blocking its active site. Int. J. Biol. Macromol. 2018, 116, 451–462. [Google Scholar] [CrossRef]
- Garmashova, N.; Gorchakov, R.; Volkova, E.; Paessler, S.; Frolova, E.; Frolov, I. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 2007, 81, 2472–2484. [Google Scholar] [CrossRef] [Green Version]
- Akhrymuk, I.; Frolov, I.; Frolova, E.I. Sindbis Virus Infection Causes Cell Death by nsP2-Induced Transcriptional Shutoff or by nsP3-Dependent Translational Shutoff. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götte, B.; Liu, L.; McInerney, G.M. The enigmatic alphavirus non-structural protein 3 (nsP3) revealing its secrets at last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, G.; Yost, S.A.; Miller, M.T.; Elrod, E.J.; Grakoui, A.; Marcotrigiano, J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 16534. [Google Scholar] [CrossRef] [Green Version]
- Tomar, S.; Hardy, R.W.; Smith, J.L.; Kuhn, R.J. Catalytic core of alphavirus nonstructural protein nsP4 possesses terminal adenylyltransferase activity. J. Virol. 2006, 80, 9962–9969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignuzzi, M.; Wendt, E.; Andino, R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 2008, 14, 154–161. [Google Scholar] [CrossRef]
- Jayabalan, A.K.; Adivarahan, S.; Koppula, A.; Abraham, R.; Batish, M.; Zenklusen, D.; Griffin, D.E.; Leung, A.K.L. Stress granule formation, disassembly, and composition are regulated by alphavirus ADP-ribosylhydrolase activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2021719118. [Google Scholar] [CrossRef] [PubMed]
- Mutso, M.; Morro, A.M.; Smedberg, C.; Kasvandik, S.; Aquilimeba, M.; Teppor, M.; Tarve, L.; Lulla, A.; Lulla, V.; Saul, S.; et al. Mutation of CD2AP and SH3KBP1 Binding Motif in Alphavirus nsP3 Hypervariable Domain Results in Attenuated Virus. Viruses 2018, 10, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemersma, K.K.; Steiner, C.; Singapuri, A.; Coffey, L.L. Chikungunya Virus Fidelity Variants Exhibit Differential Attenuation and Population Diversity in Cell Culture and Adult Mice. J. Virol. 2019, 93, e01606-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, C.M.; Liu, H.; Riemersma, K.K.; Ball, E.E.; Coffey, L.L. Engineering a fidelity-variant live-attenuated vaccine for chikungunya virus. NPJ Vaccines 2020, 5, 97. [Google Scholar] [CrossRef]
- Kautz, T.F.; Guerbois, M.; Khanipov, K.; Patterson, E.I.; Langsjoen, R.M.; Yun, R.; Warmbrod, K.L.; Fofanov, Y.; Weaver, S.C.; Forrester, N.L. Low-fidelity Venezuelan equine encephalitis virus polymerase mutants to improve live-attenuated vaccine safety and efficacy. Virus Evol. 2018, 4, vey004. [Google Scholar] [CrossRef]
- Kautz, T.F.; Forrester, N.L. RNA Virus Fidelity Mutants: A Useful Tool for Evolutionary Biology or a Complex Challenge? Viruses 2018, 10, 600. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Taylor, A. Arthritogenic Alphavirus Capsid Protein. Life 2021, 11, 230. [Google Scholar] [CrossRef]
- Geigenmüller-Gnirke, U.; Nitschko, H.; Schlesinger, S. Deletion analysis of the capsid protein of Sindbis virus: Identification of the RNA binding region. J. Virol. 1993, 67, 1620–1626. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Jose, J.; Chipman, P.; Zhang, W.; Kuhn, R.J.; Baker, T.S. Molecular links between the E2 envelope glycoprotein and nucleocapsid core in Sindbis virus. J. Mol. Biol. 2011, 414, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Rai, J.; John, L.; Schaefer, S.; Pützer, B.M.; Herchenröder, O. Chikungunya virus capsid protein contains nuclear import and export signals. Virol. J. 2013, 10, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokoloski, K.J.; Nease, L.M.; May, N.A.; Gebhart, N.N.; Jones, C.E.; Morrison, T.E.; Hardy, R.W. Identification of Interactions between Sindbis Virus Capsid Protein and Cytoplasmic vRNA as Novel Virulence Determinants. PLoS Pathog. 2017, 13, e1006473. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Zhang, W.; Gabler, S.; Chipman, P.R.; Strauss, E.G.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Mapping the Structure and Function of the E1 and E2 Glycoproteins in Alphaviruses. Structure 2006, 14, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Dropulic, L.K.; Hardwick, J.M.; Griffin, D.E. A single amino acid change in the E2 glycoprotein of Sindbis virus confers neurovirulence by altering an early step of virus replication. J. Virol. 1997, 71, 6100–6105. [Google Scholar] [CrossRef] [Green Version]
- Ryman, K.D.; Gardner, C.L.; Burke, C.W.; Meier, K.C.; Thompson, J.M.; Klimstra, W.B. Heparan sulfate binding can contribute to the neurovirulence of neuroadapted and nonneuroadapted Sindbis viruses. J. Virol. 2007, 81, 3563–3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santagati, M.G.; Määttä, J.A.; Itäranta, P.V.; Salmi, A.A.; Hinkkanen, A.E. The Semliki Forest virus E2 gene as a virulence determinant. J. Gen. Virol. 1995, 76, 47–52. [Google Scholar] [CrossRef]
- Ferguson, M.C.; Saul, S.; Fragkoudis, R.; Weisheit, S.; Cox, J.; Patabendige, A.; Sherwood, K.; Watson, M.; Merits, A.; Fazakerley, J.K. Ability of the Encephalitic Arbovirus Semliki Forest Virus to Cross the Blood-Brain Barrier Is Determined by the Charge of the E2 Glycoprotein. J. Virol. 2015, 89, 7536–7549. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Berberich, E.; von Rhein, C.; Henß, L.; Hildt, E.; Schnierle, B.S. Identification of Functional Determinants in the Chikungunya Virus E2 Protein. PLoS Negl. Trop. Dis. 2017, 11, e0005318. [Google Scholar] [CrossRef]
- McAllister, N.; Liu, Y.; Silva, L.M.; Lentscher, A.J.; Chai, W.; Wu, N.; Griswold, K.A.; Raghunathan, K.; Vang, L.; Alexander, J. Chikungunya virus strains from each genetic clade bind sulfated glycosaminoglycans as attachment factors. J. Virol. 2020, 94, e01500-20. [Google Scholar] [CrossRef] [PubMed]
- Guardado-Calvo, P.; Atkovska, K.; Jeffers, S.; Grau, N.; Backovic, M.; Pérez-Vargas, J.; De Boer, S.; Tortorici, M.A.; Pehau-Arnaudet, G.; Lepault, J. A glycerophospholipid-specific pocket in the RVFV class II fusion protein drives target membrane insertion. Science 2017, 358, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Chen, R.; Leal, G.; Forrester, N.; Higgs, S.; Huang, J.; Weaver, S.C. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc. Natl. Acad. Sci. USA 2011, 108, 7872–7877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Sharma, A.K.; Sukumaran, D.; Parida, M.; Dash, P.K. Two novel epistatic mutations (E1:K211E and E2:V264A) in structural proteins of Chikungunya virus enhance fitness in Aedes aegypti. Virology 2016, 497, 59–68. [Google Scholar] [CrossRef]
- Grandadam, M.; Caro, V.; Plumet, S.; Thiberge, J.M.; Souarès, Y.; Failloux, A.-B.; Tolou, H.J.; Budelot, M.; Cosserat, D.; Leparc-Goffart, I.; et al. Chikungunya virus, southeastern France. Emerg. Infect. Dis. 2011, 17, 910–913. [Google Scholar] [CrossRef]
- Sun, S.; Xiang, Y.; Akahata, W.; Holdaway, H.; Pal, P.; Zhang, X.; Diamond, M.S.; Nabel, G.J.; Rossmann, M.G. Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. eLife 2013, 2, e00435. [Google Scholar] [CrossRef] [PubMed]
- Voss, J.E.; Vaney, M.-C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709. [Google Scholar] [CrossRef]
- Bonatti, S.; Migliaccio, G.; Blobel, G.; WALTER, P. Role of signal recognition particle in the membrane assembly of Sindbis viral glycoproteins. Eur. J. Biochem. 1984, 140, 499–502. [Google Scholar] [CrossRef]
- Bonatti, S.; Blobel, G. Absence of a cleavable signal sequence in Sindbis virus glycoprotein PE2. J. Biol. Chem. 1979, 254, 12261–12264. [Google Scholar] [CrossRef]
- Uchime, O.; Fields, W.; Kielian, M. The role of E3 in pH protection during alphavirus assembly and exit. J. Virol. 2013, 87, 10255–10262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrott, M.M.; Sitarski, S.A.; Arnold, R.J.; Picton, L.K.; Hill, R.B.; Mukhopadhyay, S. Role of Conserved Cysteines in the Alphavirus E3 Protein. J. Virol. 2009, 83, 2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Quiñonez, J.A.; Escobar-Escamilla, N.; Ortíz-Alcántara, J.; Vázquez-Pichardo, M.; de la Luz Torres-Rodríguez, M.; Nuñez-León, A.; Torres-Longoria, B.; López-Martínez, I.; Ruiz-Matus, C.; Kuri-Morales, P. Identification of Asian genotype of chikungunya virus isolated in Mexico. Virus Genes 2016, 52, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Rodas, J.D.; Kautz, T.; Camacho, E.; Paternina, L.; Guzmán, H.; Díaz, F.J.; Blanco, P.; Tesh, R.; Weaver, S.C. Genetic characterization of northwestern Colombian chikungunya virus strains from the 2014–2015 epidemic. Am. J. Trop. Med. Hyg. 2016, 95, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Snyder, J.E.; Kulcsar, K.A.; Schultz, K.L.W.; Riley, C.P.; Neary, J.T.; Marr, S.; Jose, J.; Griffin, D.E.; Kuhn, R.J. Functional characterization of the alphavirus TF protein. J. Virol. 2013, 87, 8511–8523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, N.L.; Guerbois, M.; Adams, A.P.; Liang, X.; Weaver, S.C. Analysis of intrahost variation in Venezuelan equine encephalitis virus reveals repeated deletions in the 6-kilodalton protein gene. J. Virol. 2011, 85, 8709–8717. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, J.; Renzi, E.C.; Arnold, R.J.; Trinidad, J.C.; Mukhopadhyay, S. Palmitoylation of Sindbis Virus TF Protein Regulates Its Plasma Membrane Localization and Subsequent Incorporation into Virions. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Hyde, J.L.; Chen, R.; Trobaugh, D.W.; Diamond, M.S.; Weaver, S.C.; Klimstra, W.B.; Wilusz, J. The 5′ and 3′ ends of alphavirus RNAs–Non-coding is not non-functional. Virus Res. 2015, 206, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Kinney, R.M.; Chang, G.J.; Tsuchiya, K.R.; Sneider, J.M.; Roehrig, J.T.; Woodward, T.M.; Trent, D.W. Attenuation of Venezuelan equine encephalitis virus strain TC-83 is encoded by the 5′-noncoding region and the E2 envelope glycoprotein. J. Virol. 1993, 67, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef]
- Weaver, S.C.; Brault, A.C.; Kang, W.; Holland, J.J. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 1999, 73, 4316–4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapleford, K.A.; Moratorio, G.; Henningsson, R.; Chen, R.; Matheus, S.; Enfissi, A.; Weissglas-Volkov, D.; Isakov, O.; Blanc, H.; Mounce, B.C. Whole-genome sequencing analysis from the chikungunya virus Caribbean outbreak reveals novel evolutionary genomic elements. PLoS Negl. Trop. Dis. 2016, 10, e0004402. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, E.; Tsetsarkin, K.A.; Weaver, S.C. Chikungunya virus 3′ untranslated region: Adaptation to mosquitoes and a population bottleneck as major evolutionary forces. PLoS Pathog 2013, 9, e1003591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, V.J.; Noval, M.G.; Chen, R.; Weaver, S.C.; Vignuzzi, M.; Stapleford, K.A.; Turner, P.E. Chikungunya virus evolution following a large 3′ UTR deletion results in host-specific molecular changes in protein-coding regions. Virus Evol. 2018, 4, vey012. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Region | Mutation/Element | Description | Reference |
|---|---|---|---|
| nsP1 | I538T | Neurovirulence (SINV); IFN-I modulation (SINV, RRV) | [26,27] |
| nsP1 | S79C, L224I | Musculoskeletal inflammation (RRV) | [28] |
| nsP2 | SINV P726G, CHIKV P718S | IFN-I modulation; JAK-STAT inhibition | [29,30,31] |
| nsP2 | A31T, N219T, S580L, Q619R | RRV IFN-I modulation; RIG-I, IRF3 expression | [32] |
| nsP2 | 674ATLG677 | Transcription inhibition (CHIKV) | [33] |
| nsP2 | KR649AA, P718S | UPR inhibition (SINV) | [34] |
| nsP3 | T344, T345 | Neurovirulence (SFV) | [35] |
| nsP3 | SINV Opal-537, SFV Opal-469, CHIKV Opal-524 | Pathogenesis in mice | [36,37,38] |
| nsP3 | Residues 386–403 | Neurovirulence (SINV) | [36] |
| nsP3 | G32S/A and Y114A | Macrodomain activity, pathogenesis in mice (SINV) | [39] |
| nsP3 | G3BP-binding motif 471–483; FXR-biding motif 531–547 | Neurovirulence (EEEV) | [40] |
| nsP4 | C483Y | Fidelity, fitness in mice/mosquitoes (CHIKV) | [41] |
| nsP4 | G7R, E31G, S90T, and C482Y | Fidelity, fitness in mice/mosquitoes (VEEV) | [42] |
| nsP4 | Undefined | EIF2α phosphorylation, UPR blocking (CHIKV, SINV) | [43] |
| Capsid | N-terminal NLS | Nuclearcytoplasmic transport blocking (VEEV) | [44] |
| Capsid | L48A, F50A | Nuclear import inhibition (VEEV) | [45] |
| Capsid | Residues 55–75 | Host gene expression inhibition, IFN sensitivity, fitness in mice (EEEV) | [46] |
| Capsid | Undefined | PRR pathway inhibition (WEEV) | [47,48] |
| Capsid | K124A/K128A; K139A/K142A | RNAi blocking (SFV) | [49] |
| E1 | V72A, G313D | Neurovirulence (SINV) | [50] |
| E1 | V80I:A129V | Fitness in mice, stability (CHIKV) | [51] |
| E1 | V80L | Infectivity, dissemination in mice/mosquitoes, cholesterol dependence (CHIKV) | [52] |
| E1 | A226V | Vector tropism (CHIKV), cholesterol dependence (CHIKV, SFV, SINV) | [53,54,55,56] |
| E1 | N141Q | E1 glycosylation, clearance and IFN-γ levels in mice (RRV) | [57] |
| E2 | G55H, L209G, E70K | Neurovirulence, HS binding (SINV) | [50,58,59] |
| E2 | R82G, E166K | GAG binding, disease severity, host response (CHIKV) | [58,59,60] |
| E2 | K71A, K74A, K77A | Neurovirulence, GAG binding (EEEV) | [61] |
| E2 | K200R (CHIKV, ONNV), K251R (RRV) | Fitness and cell-mediated clearance in mice | [62,63] |
| E2 | N200Q | E2 glycosylation, mosquito infectivity (RRV) | [57] |
| E2 | N196Q, N318Q | Fitness in mice, HS binding (SINV) | [64] |
| E3 | C25R | PE2 processing (SINV) | [65] |
| 6K | Undefined | Fitness in mice (CHIKV, RRV, VEEV) | [66,67,68] |
| TF | Undefined | IFN-I antagonism | [69] |
| TF | C35, C36, C38, C39 | TF palmitoylation | [70] |
| 5′UTR | Position 5 and 8 (SINV), 21, 35, and 42 (SFV) | Fitness in mice | [71,72,73] |
| 5′UTR | G3A | IFN sensitivity, Ifit1 evasion (VEEV) | [74,75] |
| 3′UTR | Position 11,337–11,596 | miR-142-3p binding, immune detection (EEEV) | [76] |
| 3′UTR | Position 31–293 | Host adaptation, fitness in mosquitoes (SINV) | [77] |
| 3′UTR | Position 11,921–11,964 | Fitness in mice | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangel, M.V.; Stapleford, K.A. Alphavirus Virulence Determinants. Pathogens 2021, 10, 981. https://doi.org/10.3390/pathogens10080981
Rangel MV, Stapleford KA. Alphavirus Virulence Determinants. Pathogens. 2021; 10(8):981. https://doi.org/10.3390/pathogens10080981
Chicago/Turabian StyleRangel, Margarita V., and Kenneth A. Stapleford. 2021. "Alphavirus Virulence Determinants" Pathogens 10, no. 8: 981. https://doi.org/10.3390/pathogens10080981
APA StyleRangel, M. V., & Stapleford, K. A. (2021). Alphavirus Virulence Determinants. Pathogens, 10(8), 981. https://doi.org/10.3390/pathogens10080981