
Cyclophilins and Their Roles in Hepatitis C Virus and Flavivirus Infections: Perspectives for Novel Antiviral Approaches


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cyclophilins and HCV Infection
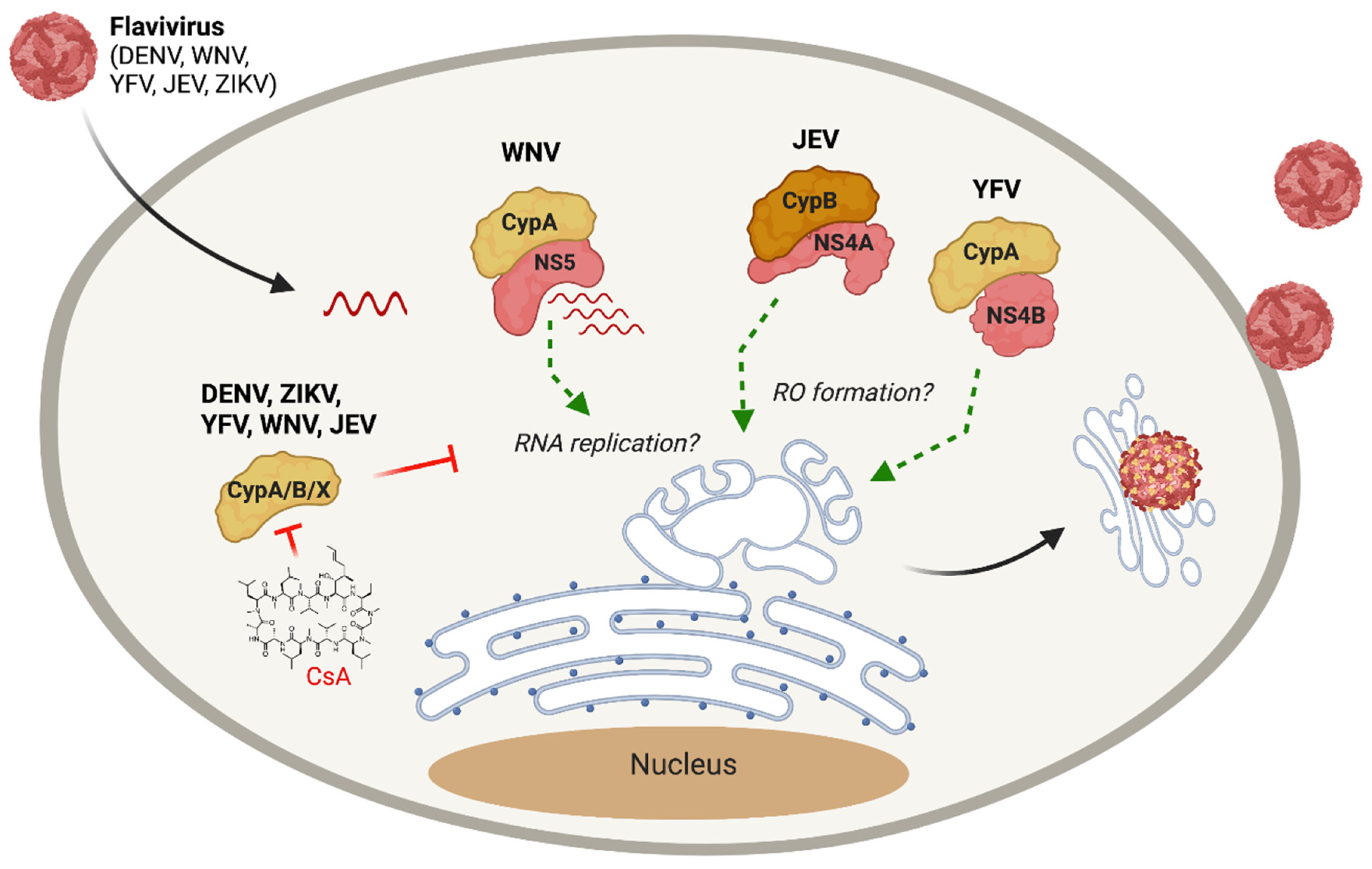
3. Cyclophilins and Other Flaviviridae
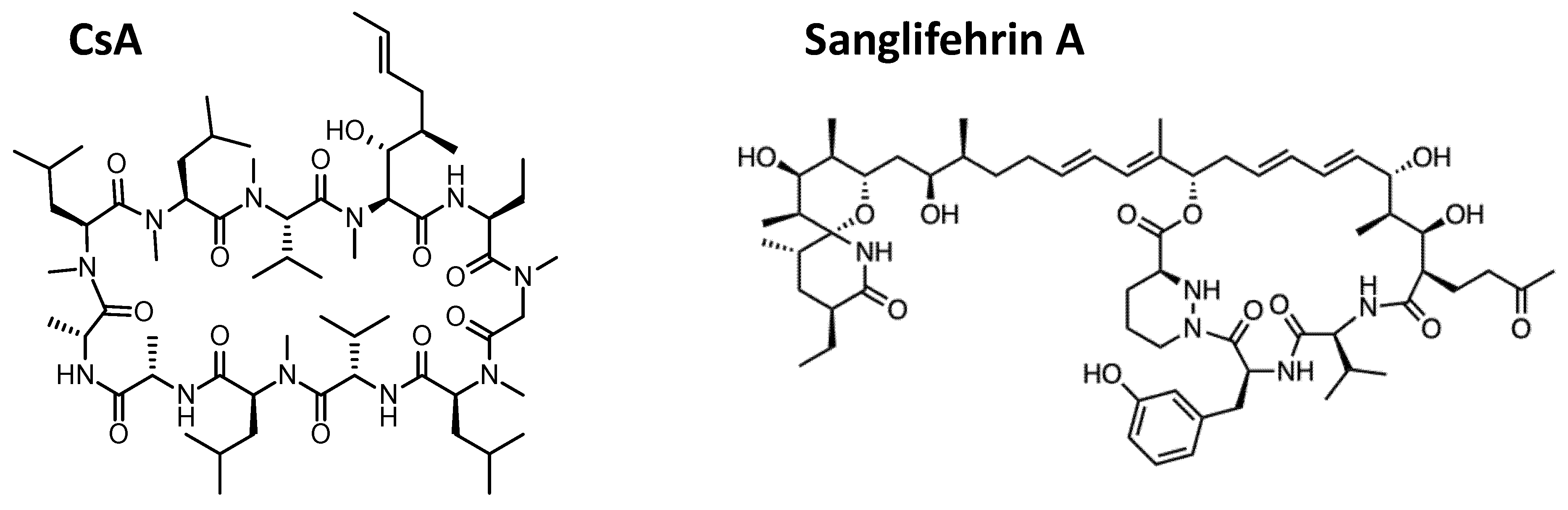
4. Future Perspectives for Cyclophilin Inhibitors as Antiviral Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef]
- Fayek, S.A.; Quintini, C.; Chavin, K.D.; Marsh, C.L. The current state of liver transplantation in the United States: Perspective from American Society of Transplant Surgeons (ASTS) scientific studies committee and endorsed by ASTS council. Am. J. Transplant. 2016. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016. [Google Scholar] [CrossRef]
- World Health Organization. Global Hepatitis Report. Available online: https://www.who.int/publications/i/item/global-hepatitis-report-2017 (accessed on 30 June 2021).
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef]
- Andrieux-Meyer, I.; Cohn, J.; de Araujo, E.S.; Hamid, S.S. Disparity in market prices for hepatitis C virus direct-acting drugs. Lancet Glob. Health 2015, 3, e676–e677. [Google Scholar] [CrossRef]
- Fourati, S.; Rodriguez, C.; Soulier, A.; Donati, F.; Hamadat, S.; Poiteau, L.; Demontant, V.; Brillet, R.; Ahnou, N.; Gricourt, G.; et al. Fitness-associated substitutions following failure of direct-acting antivirals assessed by deep sequencing of full-length hepatitis C virus genomes. Aliment. Pharm. 2020, 52, 1583–1591. [Google Scholar] [CrossRef]
- Bhatia, M.; Gupta, E. Emerging resistance to directly-acting antiviral therapy in treatment of chronic Hepatitis C infection—A brief review of literature. J. Fam. Med. Prim. Care 2020, 9, 531–538. [Google Scholar] [CrossRef]
- Colpitts, C.C.; Baumert, T.F. Addressing the Challenges of Hepatitis C Virus Resistance and Treatment Failure. Viruses 2016, 8, 226. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017. [Google Scholar] [CrossRef]
- Colpitts, C.C.; Tsai, P.L.; Zeisel, M.B. Hepatitis C virus entry: An intriguingly complex and highly regulated process. Int. J. Mol. Sci. 2020, 21, 2091. [Google Scholar] [CrossRef] [PubMed]
- Lin, C. HCV NS3-4A Serine Protease. Hepatitis C Viruses: Genomes and Molecular Biology; Horizon Bioscience: Norfolk, UK, 2006. [Google Scholar]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Joyce, M.A.; Van Buuren, N.; Levin, A.; Kirkegaard, K.; Gale, M.; Tyrrell, D.L.J.; Wozniak, R.W. The hepatitis C virus-induced membranous web and associated nuclear transport machinery limit access of pattern recognition receptors to viral replication sites. PLoS Pathog. 2016. [Google Scholar] [CrossRef]
- Kim, C.W.; Chang, K.M. Hepatitis C virus: Virology and life cycle. Clin. Mol. Hepatol. 2013. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Matsunaga, S.; Takahashi, H.; Nakashima, K.; Kimura, Y.; Ito, M.; Matsuda, M.; Murayama, A.; Kato, T.; Hirano, H.; et al. Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase I-alpha in infectious virus production. J. Virol. 2014, 88, 7541–7555. [Google Scholar] [CrossRef]
- Lindenbach, B.D. Virion assembly and release. Hepat. C Virus 2013, 369, 199–218. [Google Scholar] [CrossRef]
- Syed, G.H.; Khan, M.; Yang, S.; Siddiqui, A. Hepatitis C virus lipoviroparticles assemble in the endoplasmic reticulum (ER) and bud off from the ER to the Golgi compartment in COPII vesicles. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science 1984. [Google Scholar] [CrossRef]
- Borel, J.F.; Feurer, C.; Gubler, H.U.; Stähelin, H. Biological effects of cyclosporin A: A new antilymphocytic agent. Agents Actions 1994. [Google Scholar] [CrossRef]
- Clipstone, N.A.; Crabtree, G.R. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 1992. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991. [Google Scholar] [CrossRef]
- Goto, K.; Watashi, K.; Murata, T.; Hishiki, T.; Hijikata, M.; Shimotohno, K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 2006, 343, 879–884. [Google Scholar] [CrossRef]
- Ma, S.; Boerner, J.E.; TiongYip, C.; Weidmann, B.; Ryder, N.S.; Cooreman, M.P.; Lin, K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 2006, 50, 2976–2982. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; Scorneaux, B.; Huang, Z.; Murray, M.G.; Wring, S.; Smitley, C.; Harris, R.; Erdmann, F.; Fischer, G.; Ribeill, Y. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 2010, 54, 660–672. [Google Scholar] [CrossRef]
- Ahmed-Belkacem, A.; Colliandre, L.; Ahnou, N.; Nevers, Q.; Gelin, M.; Bessin, Y.; Brillet, R.; Cala, O.; Douguet, D.; Bourguet, W.; et al. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat. Commun. 2016, 7, 12777. [Google Scholar] [CrossRef] [PubMed]
- Mackman, R.L.; Steadman, V.A.; Dean, D.K.; Jansa, P.; Poullennec, K.G.; Appleby, T.; Austin, C.; Blakemore, C.A.; Cai, R.; Cannizzaro, C.; et al. Discovery of a potent and orally bioavailable cyclophilin inhibitor derived from the sanglifehrin macrocycle. J. Med. Chem. 2018, 61, 9473–9499. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, C.C.; Ridewood, S.; Schneiderman, B.; Warne, J.; Tabata, K.; Ng, C.F.; Bartenschlager, R.; Selwood, D.L.; Towers, G.J. Hepatitis C virus exploits cyclophilin a to evade PKR. eLife 2020. [Google Scholar] [CrossRef] [PubMed]
- Stamnes, M.A.; Rutherford, S.L.; Zuker, C.S. Cyclophilins: A new family of proteins involved in intracellular folding. Trends Cell Biol. 1992. [Google Scholar] [CrossRef]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; Mackenzie, F.; Tempel, W.; Ouyang, H.; et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010. [Google Scholar] [CrossRef]
- Kallen, J.; Spitzfaden, C.; Zurini, M.G.M.; Wider, G.; Widmer, H.; Wüthrich, K.; Walkinshaw, M.D. Structure of human cyclophilin and its binding site for cyclosporin A determined by X-ray crystallography and NMR spectroscopy. Nature 1991. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Roy, S.; Singh, P.; Singla-Pareek, S.L.; Pareek, A. Cyclophilins: Proteins in search of function. Plant. Signal. Behav. 2013. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef] [PubMed]
- Sherry, B.; Yarlett, N.; Strupp, A.; Cerami, A. Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages. Proc. Natl. Acad. Sci. USA 1992. [Google Scholar] [CrossRef]
- Spik, G.; Haendler, B.; Delmas, O.; Mariller, C.; Chamoux, M.; Maes, P.; Tartar, A.; Montreuil, J.; Stedman, K.; Kocher, H.P.; et al. A novel secreted cyclophilin-like protein (SCYLP). J. Biol. Chem. 1991. [Google Scholar] [CrossRef]
- Suzuki, J.; Jin, Z.G.; Meoli, D.F.; Matoba, T.; Berk, B.C. Cyclophilin A is secreted by a vesicular pathway in vascular smooth muscle cells. Circ. Res. 2006. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef]
- Qing, M.; Yang, F.; Zhang, B.; Zou, G.; Robida, J.M.; Yuan, Z.; Tang, H.; Shi, P.Y. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob. Agents Chemother. 2009. [Google Scholar] [CrossRef]
- Vidotto, A.; Morais, A.T.; Ribeiro, M.R.; Pacca, C.C.; Terzian, A.C.; Gil, L.H.; Mohana-Borges, R.; Gallay, P.; Nogueira, M.L. Systems biology reveals NS4B-cyclophilin A interaction: A new target to inhibit YFV replication. J. Proteome Res. 2017, 16, 1542–1555. [Google Scholar] [CrossRef]
- Barrows, N.J.; Campos, R.K.; Powell, S.T.; Prasanth, K.R.; Schott-Lerner, G.; Soto-Acosta, R.; Galarza-Munoz, G.; McGrath, E.L.; Urrabaz-Garza, R.; Gao, J.; et al. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe 2016, 20, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Tani, H.; Mori, Y.; Abe, T.; Katoh, H.; Fukuhara, T.; Taguwa, S.; Moriishi, K.; Matsuura, Y. Involvement of cyclophilin B in the replication of Japanese encephalitis virus. Virology 2011, 412, 211–219. [Google Scholar] [CrossRef]
- Pfefferle, S.; Schöpf, J.; Kögl, M.; Friedel, C.C.; Müller, M.A.; Carbajo-Lozoya, J.; Stellberger, T.; von Dall’Armi, E.; Herzog, P.; Kallies, S.J.P.p. The SARS-coronavirus-host interactome: Identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 2011, 7, e1002331. [Google Scholar] [CrossRef] [PubMed]
- Carbajo-Lozoya, J.; Ma-Lauer, Y.; Malesevic, M.; Theuerkorn, M.; Kahlert, V.; Prell, E.; von Brunn, B.; Muth, D.; Baumert, T.F.; Drosten, C.; et al. Human coronavirus NL63 replication is cyclophilin A-dependent and inhibited by non-immunosuppressive cyclosporine A-derivatives including Alisporivir. Virus Res. 2014, 184, 44–53. [Google Scholar] [CrossRef] [PubMed]
- de Wilde, A.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Thiel, V.; Narayanan, K.; Makino, S.; Snijder, E.J.; van Hemert, M.J. Cyclosporin A inhibits the replication of diverse coronaviruses. J. Gen. Virol. 2011, 92, 2542–2548. [Google Scholar] [CrossRef] [PubMed]
- de Wilde, A.H.; Zevenhoven-Dobbe, J.C.; Beugeling, C.; Chatterji, U.; de Jong, D.; Gallay, P.; Szuhai, K.; Posthuma, C.C.; Snijder, E.J. Coronaviruses and arteriviruses display striking differences in their cyclophilin A-dependence during replication in cell culture. Virology 2018, 517, 148–156. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Zheng, Y.; Malesevic, M.; von Brunn, B.; Fischer, G.; von Brunn, A. Influences of cyclosporin A and non-immunosuppressive derivatives on cellular cyclophilins and viral nucleocapsid protein during human coronavirus 229E replication. Antivir. Res. 2020, 173, 104620. [Google Scholar] [CrossRef] [PubMed]
- Sauerhering, L.; Kupke, A.; Meier, L.; Dietzel, E.; Hoppe, J.; Gruber, A.D.; Gattenloehner, S.; Witte, B.; Fink, L.; Hofmann, N.; et al. Cyclophilin inhibitors restrict Middle East respiratory syndrome coronavirus via interferon-lambda in vitro and in mice. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Softic, L.; Brillet, R.; Berry, F.; Ahnou, N.; Nevers, Q.; Morin-Dewaele, M.; Hamadat, S.; Bruscella, P.; Fourati, S.; Pawlotsky, J.M.; et al. Inhibition of SARS-CoV-2 infection by the cyclophilin inhibitor alisporivir (Debio 025). Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Watashi, K.; Shimotohno, K. Cyclophilin and viruses: Cyclophilin as a cofactor for viral infection and possible anti-viral target. Drug Target. Insights 2007, 2, 9–18. [Google Scholar] [CrossRef]
- Watashi, K.; Hijikata, M.; Hosaka, M.; Yamaji, M.; Shimotohno, K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 2003. [Google Scholar] [CrossRef]
- Nakagawa, M.; Sakamoto, N.; Enomoto, N.; Tanabe, Y.; Kanazawa, N.; Koyama, T.; Kurosaki, M.; Maekawa, S.; Yamashiro, T.; Chen, C.H.; et al. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 2004, 313, 42–47. [Google Scholar] [CrossRef]
- Ishii, N.; Watashi, K.; Hishiki, T.; Goto, K.; Inoue, D.; Hijikata, M.; Wakita, T.; Kato, N.; Shimotohno, K. Diverse effects of cyclosporine on hepatitis C virus strain replication. J. Virol. 2006, 80, 4510–4520. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Sekiyama, K.; Yamada, M.; Watanabe, T.; Yasuda, H.; Yoshiba, M. Combined interferon α2b and cyclosporin A in the treatment of chronic hepatitis C: Controlled trial. J. Gastroenterol. 2003. [Google Scholar] [CrossRef]
- Inoue, K.; Yoshiba, M. Interferon combined with cyclosporine treatment as an effective countermeasure against hepatitis C virus recurrence in liver transplant patients with end-stage hepatitis C virus related disease. Transplant. Proc. 2005, 37, 1233–1234. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, C.; Trifan, A.; Muzica, C.; Sfarti, C. Efficacy and safety of alisporivir for the treatment of hepatitis C infection. Expert Opin. Pharmacother. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; Gallay, P. Cyclophilin inhibitors: An emerging class of therapeutics for the treatment of chronic hepatitis C infection. Viruses 2012, 4, 2558–2577. [Google Scholar] [CrossRef]
- Nakagawa, M.; Sakamoto, N.; Tanabe, Y.; Koyama, T.; Itsui, Y.; Takeda, Y.; Chen, C.H.; Kakinuma, S.; Oooka, S.; Maekawa, S.; et al. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology 2005. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Ishii, N.; Hijikata, M.; Inoue, D.; Murata, T.; Miyanari, Y.; Shimotohno, K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 2005. [Google Scholar] [CrossRef]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008. [Google Scholar] [CrossRef]
- Chatterji, U.; Bobardt, M.; Selvarajah, S.; Yang, F.; Tang, H.; Sakamoto, N.; Vuagniaux, G.; Parkinson, T.; Gallay, P. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Lopez, M.Z.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009. [Google Scholar] [CrossRef]
- Fernandes, F.; Poole, D.S.; Hoover, S.; Middleton, R.; Andrei, A.C.; Gerstner, J.; Striker, R. Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology 2007. [Google Scholar] [CrossRef] [PubMed]
- Heck, J.A.; Meng, X.; Frick, D.N. Cyclophilin B stimulates RNA synthesis by the HCV RNA dependent RNA polymerase. Biochem. Pharmacol. 2009. [Google Scholar] [CrossRef]
- Weng, L.; Tian, X.; Gao, Y.; Watashi, K.; Shimotohno, K.; Wakita, T.; Kohara, M.; Toyoda, T. Different mechanisms of hepatitis C virus RNA polymerase activation by cyclophilin A and B in vitro. Biochim. Et Biophys. Acta-Gen. Subj. 2012, 1820, 1886–1892. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J.C.; Rice, C.M. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 2008. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Hanoulle, X.; Badillo, A.; Wieruszeski, J.M.; Verdegem, D.; Landrieu, I.; Bartenschlager, R.; Penin, F.; Lippens, G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 2009, 284, 13589–13601. [Google Scholar] [CrossRef]
- Chatterji, U.; Lim, P.; Bobardt, M.D.; Wieland, S.; Cordek, D.G.; Vuagniaux, G.; Chisari, F.; Cameron, C.E.; Targett-Adams, P.; Parkinson, T.; et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 2010. [Google Scholar] [CrossRef]
- Foster, T.L.; Gallay, P.; Stonehouse, N.J.; Harris, M. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Grise, H.; Frausto, S.; Logan, T.; Tang, H. A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates Alisporivir susceptibility. J. Virol. 2012, 86, 4811–4822. [Google Scholar] [CrossRef]
- Coelmont, L.; Hanoulle, X.; Chatterji, U.; Berger, C.; Snoeck, J.; Bobardt, M.; Lim, P.; Vliegen, I.; Paeshuyse, J.; Vuagniaux, G.; et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS ONE 2010, 5, e13687. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Robotham, J.M.; Grise, H.; Frausto, S.; Madan, V.; Zayas, M.; Bartenschlager, R.; Robinson, M.; Greenstein, A.E.; Nag, A.; et al. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 2010, 6, e1001118. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; Robotham, J.M.; Tang, H. Suppression of viral RNA binding and the assembly of infectious hepatitis C virus particles in vitro by cyclophilin inhibitors. J. Virol. 2012, 86, 12616–12624. [Google Scholar] [CrossRef]
- Dujardin, M.; Madan, V.; Gandhi, N.S.; Cantrelle, F.X.; Launay, H.; Huvent, I.; Bartenschlager, R.; Lippens, G.; Hanoulle, X. Cyclophilin A allows the allosteric regulation of a structural motif in the disordered domain 2 of NS5A and thereby fine-tunes HCV RNA replication. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Rosnoblet, C.; Fritzinger, B.; Legrand, D.; Launay, H.e.; Wieruszeski, J.M.; Lippens, G.; Hanoulle, X. Hepatitis C virus NS5B and host cyclophilin A share a common binding site on NS5A. J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- Ngure, M.; Issur, M.; Shkriabai, N.; Liu, H.W.; Cosa, G.; Kvaratskhelia, M.; Götte, M. Interactions of the disordered domain II of hepatitis C virus NS5A with cyclophilin A, NS5B, and viral RNA show extensive overlap. ACS Infect. Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, F.; Robotham, J.M.; Tang, H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 2009, 83, 6554–6565. [Google Scholar] [CrossRef]
- Madan, V.; Paul, D.; Lohmann, V.; Bartenschlager, R. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 2014. [Google Scholar] [CrossRef]
- Chatterji, U.; Bobardt, M.; Tai, A.; Wood, M.; Gallay, P.A. Cyclophilin and NS5A inhibitors, but not other anti-hepatitis C virus (HCV) agents, preclude HCV-mediated formation of double-membrane-vesicle viral factories. Antimicrob. Agents Chemother. 2015, 59, 2496–2507. [Google Scholar] [CrossRef]
- Ciesek, S.; Steinmann, E.; Wedemeyer, H.; Manns, M.P.; Neyts, J.; Tautz, N.; Madan, V.; Bartenschlager, R.; von Hahn, T.; Pietschmann, T. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology 2009, 50, 1638–1645. [Google Scholar] [CrossRef]
- Verdegem, D.; Badillo, A.; Wieruszeski, J.M.; Landrieu, I.; Leroy, A.; Bartenschlager, R.; Penin, F.; Lippens, G.; Hanoulle, X. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J. Biol. Chem. 2011, 286, 20441–20454. [Google Scholar] [CrossRef]
- Anderson, L.J.; Lin, K.; Compton, T.; Wiedmann, B. Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 2011, 8, 329. [Google Scholar] [CrossRef]
- Daito, T.; Watashi, K.; Sluder, A.; Ohashi, H.; Nakajima, S.; Borroto-Esoda, K.; Fujita, T.; Wakita, T. Cyclophilin inhibitors reduce phosphorylation of RNA-dependent protein kinase to restore expression of IFN-stimulated genes in HCV-infected cells. Gastroenterology 2014. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Chatterji, U.; Lim, P.; Gawlik, K.; Gallay, P. Both cyclophilin inhibitors and direct-acting antivirals prevent PKR activation in HCV-infected cells. Open Virol. J. 2014, 8, 1–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pflugheber, J.; Fredericksen, B.; Sumpter, R., Jr.; Wang, C.; Ware, F.; Sodora, D.L.; Gale, M., Jr. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc. Natl. Acad. Sci. USA 2002, 99, 4650–4655. [Google Scholar] [CrossRef] [PubMed]
- Gale, M., Jr.; Blakely, C.M.; Kwieciszewski, B.; Tan, S.L.; Dossett, M.; Tang, N.M.; Korth, M.J.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: Molecular mechanisms of kinase regulation. Mol. Cell. Biol. 1998, 18, 5208–5218. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Halstead, S.B. Dengue. Lancet 2007, 370, 1644–1652. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013. [Google Scholar] [CrossRef]
- Petersen, L.R.; Hayes, E.B. West Nile virus in the Americas. Med. Clin. N. Am. 2008, 92, 1307–1322. [Google Scholar] [CrossRef]
- Shukla, R.; Ramasamy, V.; Shanmugam, R.K.; Ahuja, R.; Khanna, N. Antibody-dependent enhancement: A challenge for developing a safe dengue vaccine. Front. Cell. Infect. Microbiol. 2020, 10, 572681. [Google Scholar] [CrossRef]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Gómez, M.M.; Dos Santos, A.A.C.; De Abreu, F.V.S.; Ferreira-de-Brito, A.; De Miranda, R.M.; De Castro, M.G.; Lourenço-de-Oliveira, R. Genome analysis of yellow fever virus of the ongoing outbreak in brazil reveals polymorphisms. Mem. Inst. Oswaldo Cruz 2017. [Google Scholar] [CrossRef] [PubMed]
- Sahili, E.A.; Lescar, J. Dengue virus non-structural protein 5. Viruses 2017, 9, 91. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Nevers, Q.; Ruiz, I.; Ahnou, N.; Donati, F.; Brillet, R.; Softic, L.; Chazal, M.; Jouvenet, N.; Fourati, S.; Baudesson, C.; et al. Characterization of the anti-hepatitis C virus activity of new nonpeptidic small-molecule cyclophilin inhibitors with the potential for broad anti-Flaviviridae activity. Antimicrob. Agents Chemother. 2018. [Google Scholar] [CrossRef]
- Deviatkin, A.A.; Kholodilov, I.S.; Vakulenko, Y.A.; Karganova, G.G.; Lukashev, A.N. Tick-borne encephalitis virus: An emerging ancient zoonosis? Viruses 2020, 12, 247. [Google Scholar] [CrossRef] [PubMed]
- Tambo, E.; El-Dessouky, A.G. Defeating re-emerging Alkhurma hemorrhagic fever virus outbreak in Saudi Arabia and worldwide. PLoS Negl. Trop. Dis. 2018, 12, e0006707. [Google Scholar] [CrossRef] [PubMed]
- Hermance, M.E.; Thangamani, S. Powassan virus: An emerging arbovirus of public health concern in North America. Vector-Borne Zoonotic Dis. 2017, 17, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Bobardt, M.; Obeid, S.; Chatterji, U.; Coates, N.J.; Foster, T.; Gallay, P.; Leyssen, P.; Moss, S.J.; Neyts, J.; et al. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob. Agents Chemother. 2011, 55, 1975–1981. [Google Scholar] [CrossRef]
- Bobardt, M.; Hansson, M.J.; Mayo, P.; Ure, D.; Foster, R.; Gallay, P. Structurally distinct cyclosporin and sanglifehrin analogs CRV431 and NV556 suppress established HCV infection in humanized-liver mice. PLoS ONE 2020, 15, e0237236. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallardo-Flores, C.E.; Colpitts, C.C. Cyclophilins and Their Roles in Hepatitis C Virus and Flavivirus Infections: Perspectives for Novel Antiviral Approaches. Pathogens 2021, 10, 902. https://doi.org/10.3390/pathogens10070902
Gallardo-Flores CE, Colpitts CC. Cyclophilins and Their Roles in Hepatitis C Virus and Flavivirus Infections: Perspectives for Novel Antiviral Approaches. Pathogens. 2021; 10(7):902. https://doi.org/10.3390/pathogens10070902
Chicago/Turabian StyleGallardo-Flores, Carla E., and Che C. Colpitts. 2021. "Cyclophilins and Their Roles in Hepatitis C Virus and Flavivirus Infections: Perspectives for Novel Antiviral Approaches" Pathogens 10, no. 7: 902. https://doi.org/10.3390/pathogens10070902
APA StyleGallardo-Flores, C. E., & Colpitts, C. C. (2021). Cyclophilins and Their Roles in Hepatitis C Virus and Flavivirus Infections: Perspectives for Novel Antiviral Approaches. Pathogens, 10(7), 902. https://doi.org/10.3390/pathogens10070902