
Single-Agent and Fixed-Dose Combination HIV-1 Protease Inhibitor Drugs in Fission Yeast (Schizosaccharomyces pombe)

 ,
, 
Abstract
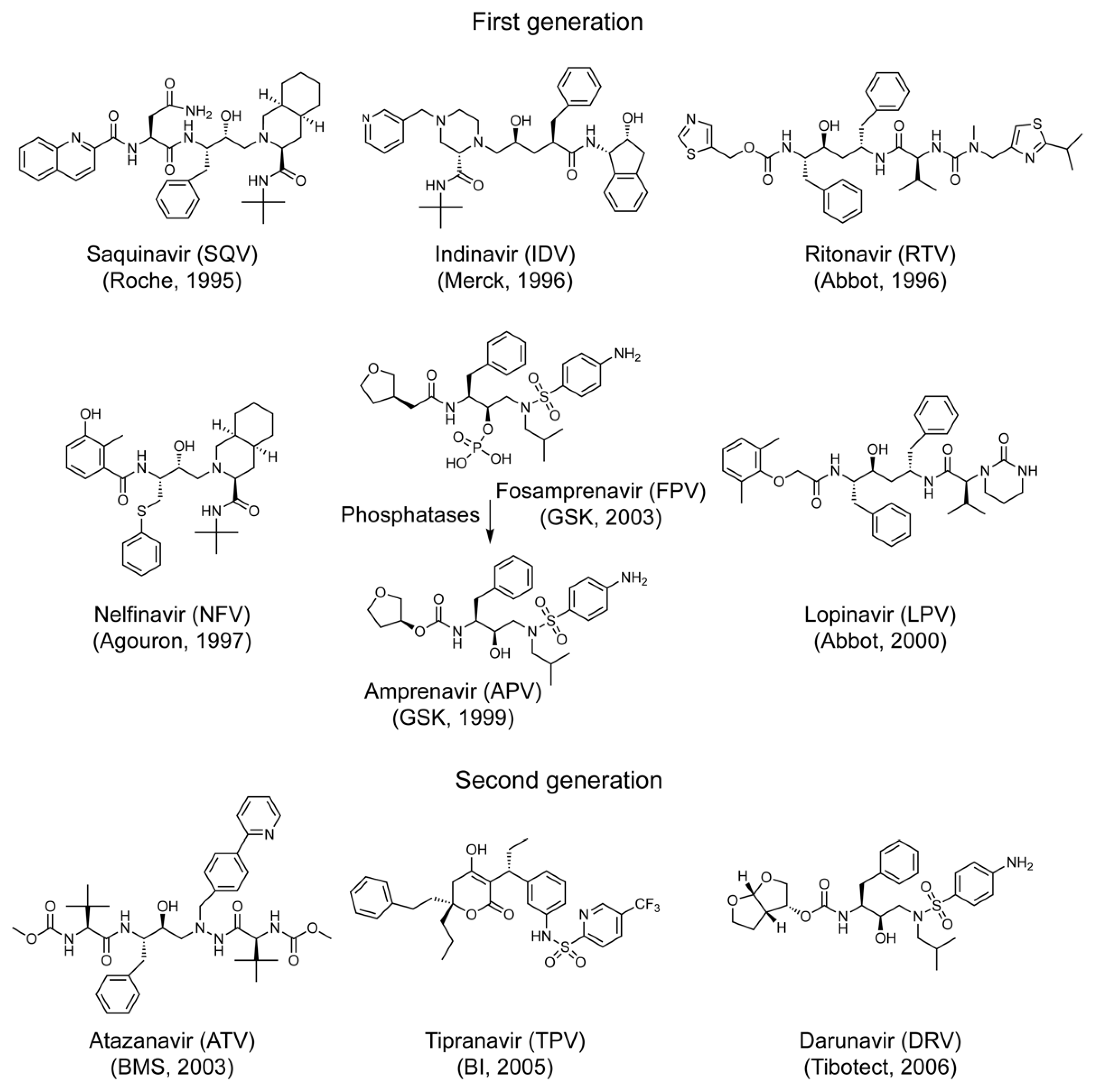
1. Introduction
2. Results
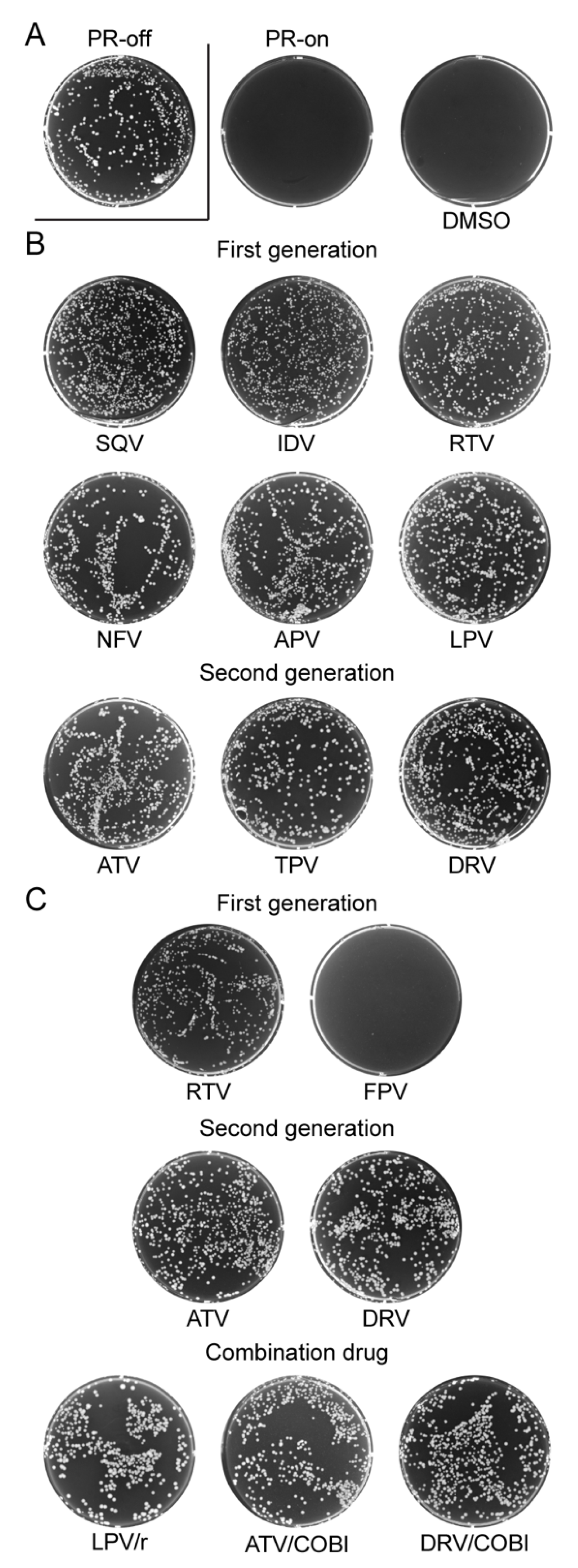
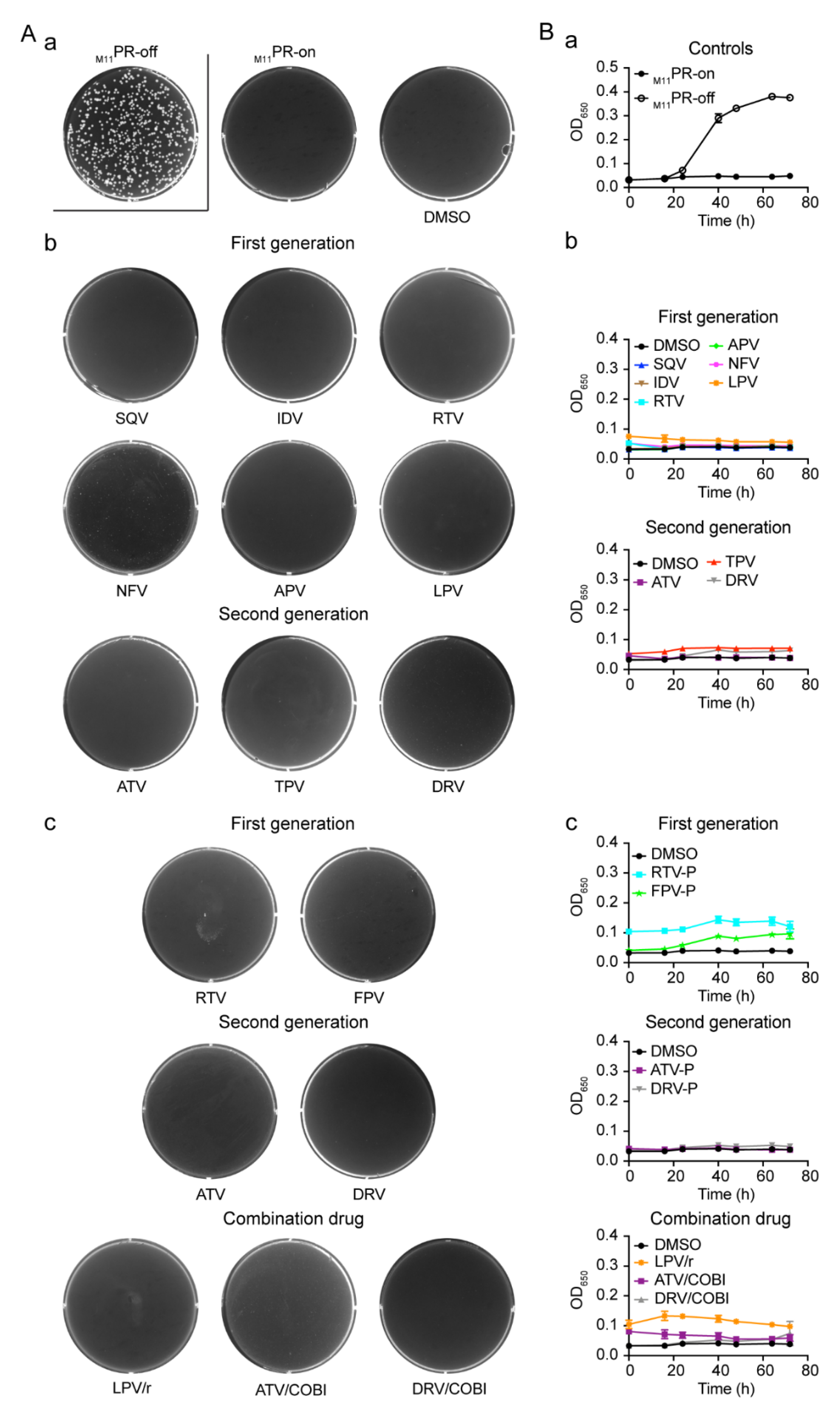
2.1. Effect of FDA-Approved Protease Inhibitor Drugs on HIV-1 Protease Measured by Colony Formation of Fission Yeast Cells
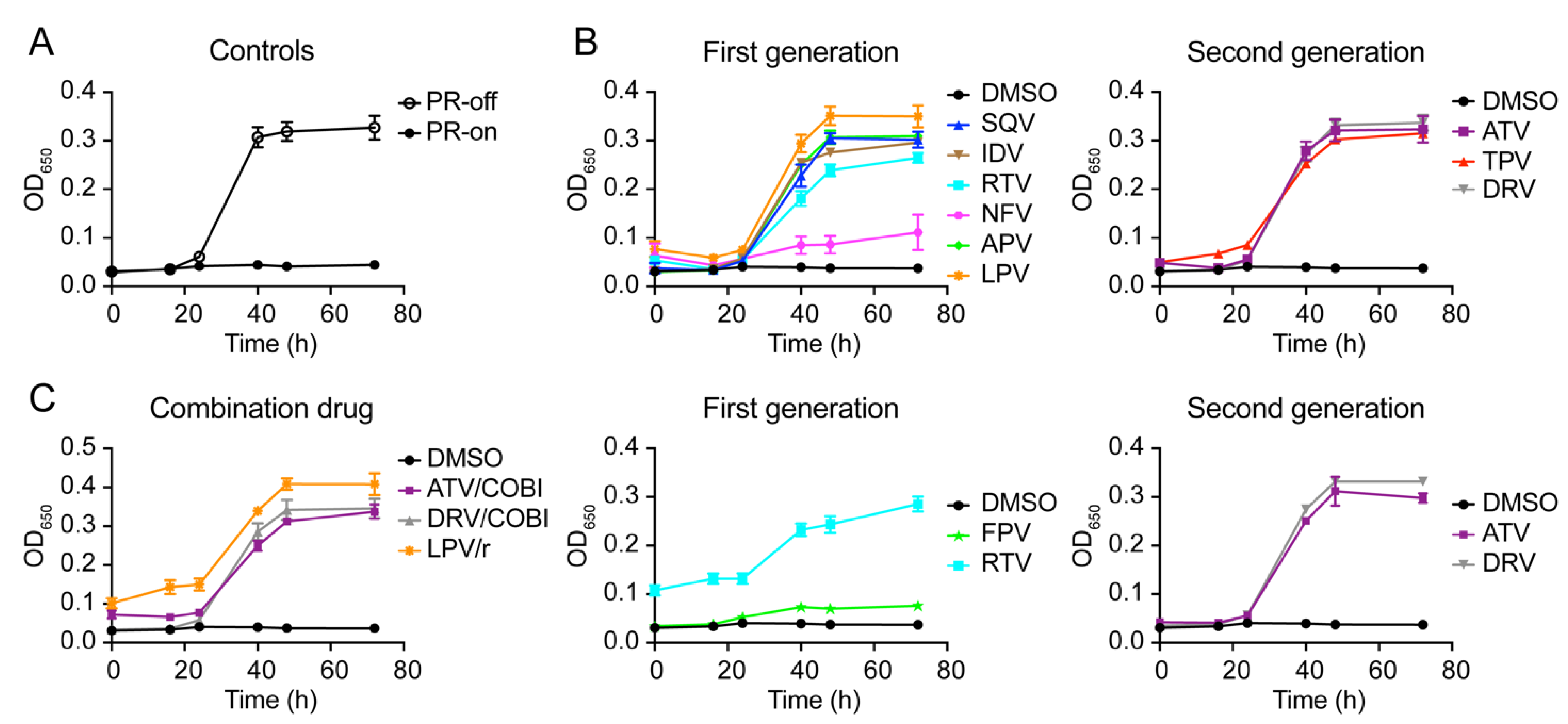
2.2. Effect of FDA-Approved Protease Inhibitor Drugs on HIV-1 Protease-Induced Growth Arrest
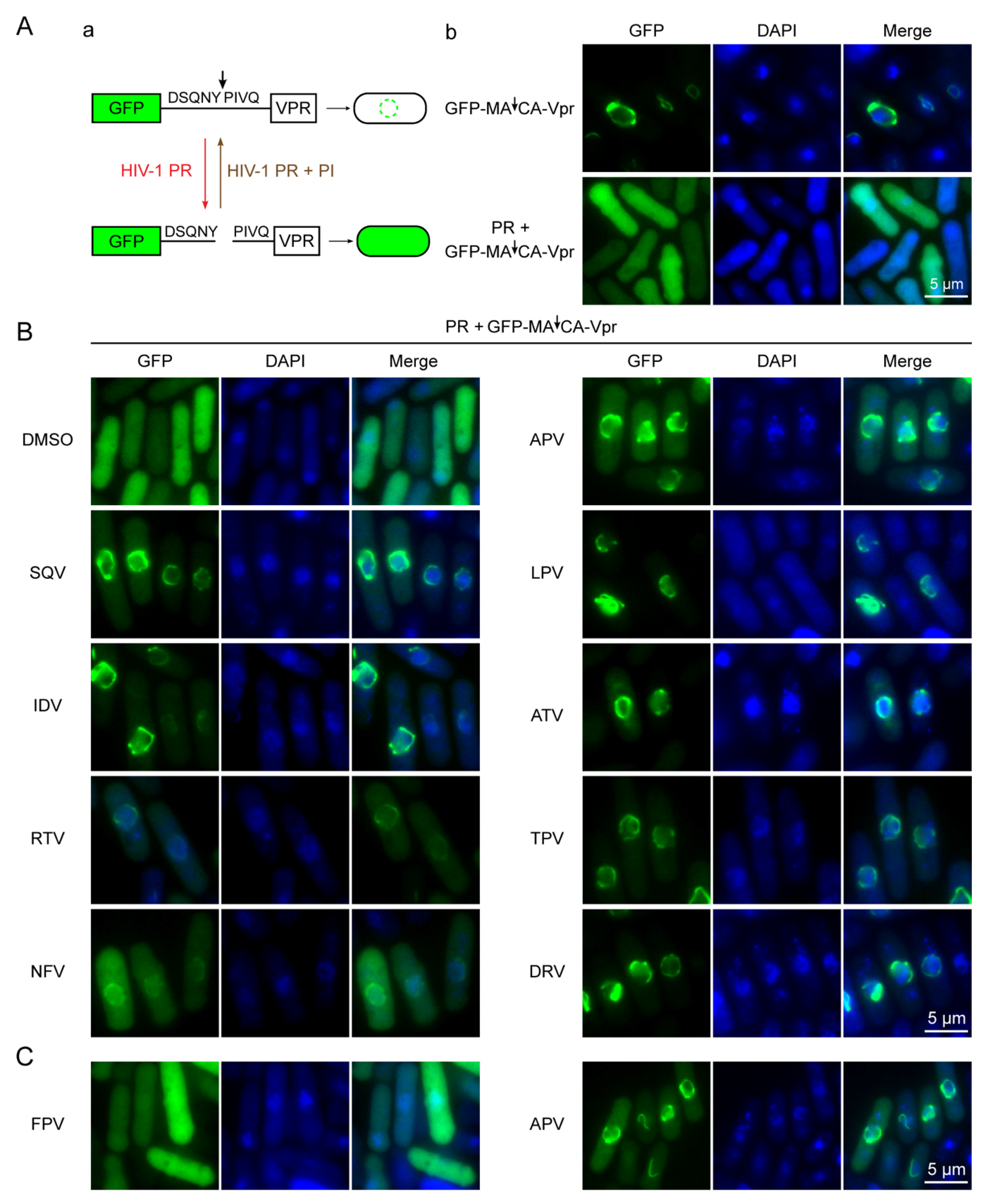
2.3. Effect of FDA-Approved Protease Inhibitor Drugs on Substrate Specific Proteolytic Cleavages of HIV-1 Protease in Fission Yeast
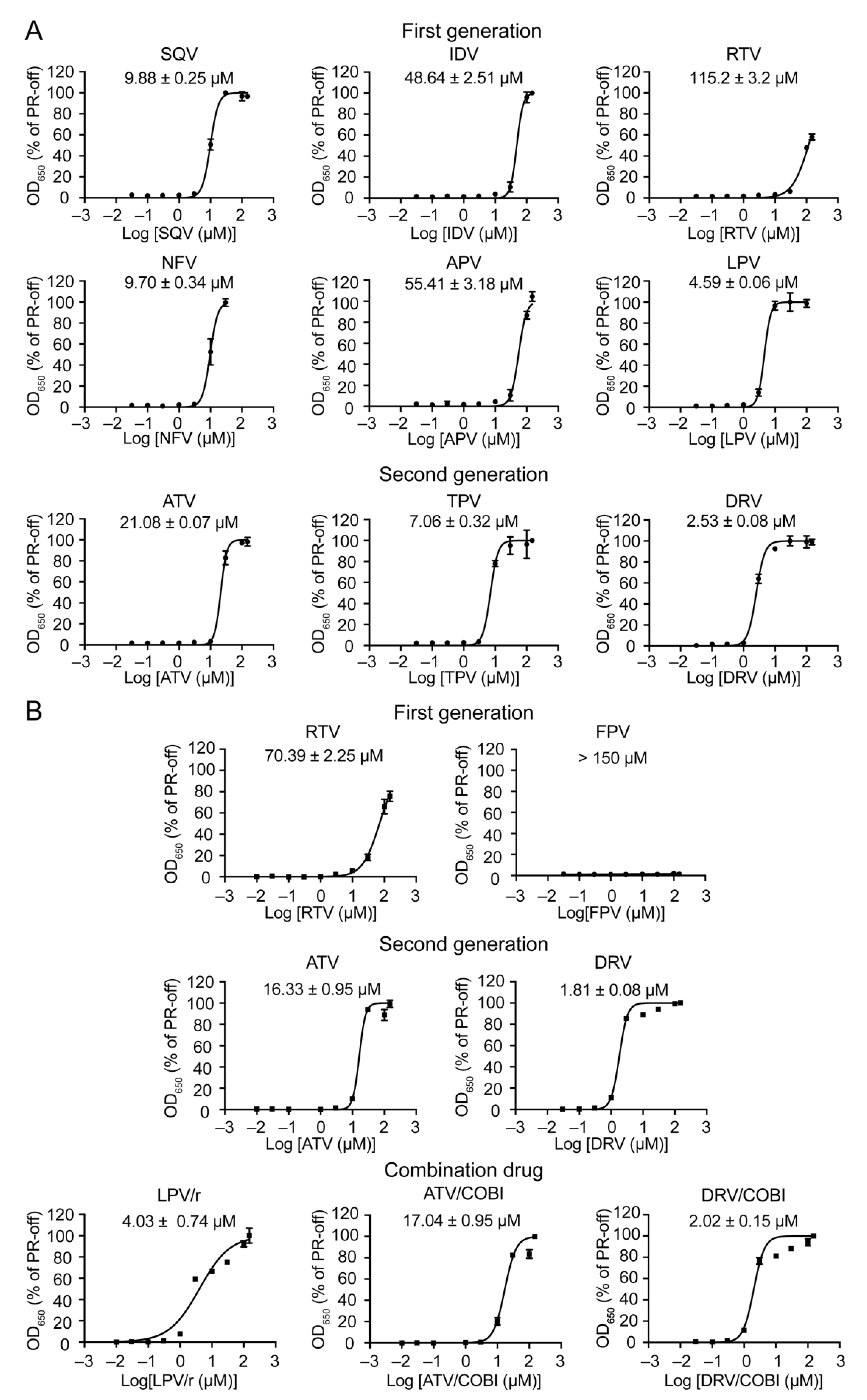
2.4. Determination of Half Maximal Effective Concentration of FDA-Approved PI Drugs in Fission Yeast
2.5. Effect of FDA-Approved Protease Inhibitor Drugs on a Multi-PI Drug Resistant HIV-1 Protease (M11PR) in Fission Yeast
3. Discussion
4. Materials and Methods
4.1. Fission Yeast Strains and Inducible Gene Expression
4.2. FDA-Approved HIV Protease Inhibitor Drugs
4.3. Fission Yeast Colony Formation Assay
4.4. Fission Yeast Growth Assay
4.5. Measurement of HIV PR Enzymatic Activity in Fission Yeast
4.6. Measurement of Half Maximal Effective Concentration of FDA-Approved PI Drugs in Fission Yeast
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jackson, S.S.; Sumner, L.E.; Finnegan, M.A.; Billings, E.A.; Huffman, D.L.; Rush, M.A. A 35-year review of pre-clinical HIV therapeutics research reported by NIH ChemDB: Influences of target discoveries, drug approvals and research funding. J. AIDS Clin. Res. 2020, 11, 11. [Google Scholar]
- Markham, A. Fostemsavir: First approval. Drugs 2020, 80, 1485–1490. [Google Scholar] [CrossRef]
- Bhatti, A.B.; Usman, M.; Kandi, V. Current scenario of HIV/AIDS, treatment options, and major challenges with compliance to antiretroviral therapy. Cureus 2016, 8, e515. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Nkeze, J.; Zhao, R.Y. Effects of HIV-1 protease on cellular functions and their potential applications in antiretroviral therapy. Cell Biosci. 2012, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Calugi, C.; Guarna, A.; Trabocchi, A. Heterocyclic HIV-protease inhibitors. Curr. Med. Chem. 2013, 20, 3693–3710. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Liu, Z.; Zhai, G. Advances in non-peptidomimetic HIV protease inhibitors. Curr. Med. Chem. 2014, 21, 1997–2011. [Google Scholar] [CrossRef] [PubMed]
- Mallolas, J. Darunavir stands up as preferred HIV protease inhibitor. AIDS Rev. 2017, 19, 105–112. [Google Scholar] [PubMed]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef]
- Persaud, D.; Siberry, G.K.; Ahonkhai, A.; Kajdas, J.; Monie, D.; Hutton, N.; Watson, D.C.; Quinn, T.C.; Ray, S.C.; Siliciano, R.F. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J. Virol. 2004, 78, 968–979. [Google Scholar] [CrossRef]
- Combescure, C.; Vallier, N.; Ledergerber, B.; Cavassini, M.; Furrer, H.; Rauch, A.; Battegay, M.; Bernasconi, E.; Vernazza, P.; Hirschel, B.; et al. How reliable is an undetectable viral load? HIV Med. 2009, 10, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Thompson, W.J.; McKee, S.P.; Duong, T.T.; Lyle, T.A.; Chen, J.C.; Darke, P.L.; Zugay, J.A.; Emini, E.A.; Schleif, W.A.; et al. 3-Tetrahydrofuran and pyran urethanes as high-affinity P2-ligands for HIV-1 protease inhibitors. J. Med. Chem. 1993, 36, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Brunton, L.L.; Lazo, J.S.; Parker, K.L. Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 11th ed.; McGraw-Hill: New York, NY, USA, 2006. [Google Scholar]
- Laco, G.S.; Schalk-Hihi, C.; Lubkowski, J.; Morris, G.; Zdanov, A.; Olson, A.; Elder, J.H.; Wlodawer, A.; Gustchina, A. Crystal structures of the inactive D30N mutant of feline immunodeficiency virus protease complexed with a substrate and an inhibitor. Biochemistry 1997, 36, 10696–10708. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Anderson, D.D.; Weber, I.T.; Mitsuya, H. Enhancing protein backbone binding—A fruitful concept for combating drug-resistant HIV. Angew. Chem. Int. Ed. 2012, 51, 1778–1802. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Darunavir: A review of its use in the management of HIV-1 infection. Drugs 2014, 74, 99–125. [Google Scholar] [CrossRef] [PubMed]
- Zeldin, R.K.; Petruschke, R.A. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J. Antimicrob. Chemother. 2004, 53, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.M.; Gibiansky, L.; Wire, M.B. Population pharmacokinetic modeling and simulation of amprenavir following fosamprenavir/ritonavir administration for dose optimization in HIV infected pediatric patients. J. Clin. Pharmacol. 2014, 54, 206–214. [Google Scholar] [CrossRef]
- Palmer, S.; Shafer, R.W.; Merigan, T.C. Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS 1999, 13, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, B.C.; Vickrey, J.F.; Martin, P.; Proteasa, G.; Koepke, J.I.; Terlecky, S.R.; Wawrzak, Z.; Winters, M.A.; Merigan, T.C.; Kovari, L.C. Crystal structures of a multidrug-resistant human immunodeficiency virus type 1 protease reveal an expanded active-site cavity. J. Virol. 2004, 78, 3123–3132. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Taylor, J.; Fessel, W.J.; Kaufman, D.; Towner, W.; Troia, P.; Ruane, P.; Hellinger, J.; Shirvani, V.; Zolopa, A.; et al. HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrob. Agents Chemother. 2010, 54, 4253–4261. [Google Scholar] [CrossRef]
- Ssemwanga, D.; Lihana, R.W.; Ugoji, C.; Abimiku, A.; Nkengasong, J.; Dakum, P.; Ndembi, N. Update on HIV-1 acquired and transmitted drug resistance in Africa. AIDS Rev. 2015, 17, 3–20. [Google Scholar] [PubMed]
- Tang, M.W.; Shafer, R.W. HIV-1 antiretroviral resistance: Scientific principles and clinical applications. Drugs 2012, 72, e1–e25. [Google Scholar] [CrossRef] [PubMed]
- Kozyryev, I.; Zhang, J. Bayesian analysis of complex interacting mutations in HIV drug resistance and cross-resistance. Adv. Exp. Med. Biol. 2015, 827, 367–383. [Google Scholar] [CrossRef]
- Benko, Z.; Elder, R.T.; Li, G.; Liang, D.; Zhao, R.Y. HIV-1 Protease in the Fission Yeast Schizosaccharomyces pombe. PLoS ONE 2016, 11, e0151286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanchez, P.R.; Holguin, A. Drug resistance in the HIV-1-infected paediatric population worldwide: A systematic review. J. Antimicrob. Chemother. 2014, 69, 2032–2042. [Google Scholar] [CrossRef]
- Cote, H.C.; Brumme, Z.L.; Harrigan, P.R. Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J. Virol. 2001, 75, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Schiffer, C.A.; Gonzales, M.J.; Taylor, J.; Kantor, R.; Chou, S.; Israelski, D.; Zolopa, A.R.; Fessel, W.J.; Shafer, R.W. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J. Virol. 2003, 77, 4836–4847. [Google Scholar] [CrossRef] [PubMed]
- Kantor, R.; Fessel, W.J.; Zolopa, A.R.; Israelski, D.; Shulman, N.; Montoya, J.G.; Harbour, M.; Schapiro, J.M.; Shafer, R.W. Evolution of primary protease inhibitor resistance mutations during protease inhibitor salvage therapy. Antimicrob. Agents Chemother. 2002, 46, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Voshavar, C. Protease inhibitors for the treatment of HIV/AIDS: Recent advances and future challenges. Curr. Top. Med. Chem. 2019, 19, 1571–1598. [Google Scholar] [CrossRef]
- Vickrey, J.F.; Logsdon, B.C.; Proteasa, G.; Palmer, S.; Winters, M.A.; Merigan, T.C.; Kovari, L.C. HIV-1 protease variants from 100-fold drug resistant clinical isolates: Expression, purification, and crystallization. Protein Expr. Purif. 2003, 28, 165–172. [Google Scholar] [CrossRef]
- Benko, Z.; Liang, D.; Li, G.; Elder, R.T.; Sarkar, A.; Takayama, J.; Ghosh, A.K.; Zhao, R.Y. A fission yeast cell-based system for multidrug resistant HIV-1 proteases. Cell Biosci. 2017, 7, 5. [Google Scholar] [CrossRef]
- Koh, Y.; Aoki, M.; Danish, M.L.; Aoki-Ogata, H.; Amano, M.; Das, D.; Shafer, R.W.; Ghosh, A.K.; Mitsuya, H. Loss of protease dimerization inhibition activity of darunavir is associated with the acquisition of resistance to darunavir by HIV-1. J. Virol. 2011, 85, 10079–10089. [Google Scholar] [CrossRef]
- Aoki, M.; Das, D.; Hayashi, H.; Aoki-Ogata, H.; Takamatsu, Y.; Ghosh, A.K.; Mitsuya, H. Mechanism of darunavir (DRV)’s high genetic barrier to HIV-1 resistance: A key V32I substitution in protease rarely occurs, but once it occurs, it predisposes HIV-1 to develop DRV resistance. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Agniswamy, J.; Harrison, R.W.; Weber, I.T. Highly drug-resistant HIV-1 protease reveals decreased intra-subunit interactions due to clusters of mutations. FEBS J. 2020, 287, 3235–3254. [Google Scholar] [CrossRef] [PubMed]
- Benko, Z.; Zhang, J.; Zhao, R.Y. Development of A fission yeast cell-based platform for high throughput screening of HIV-1 protease inhibitors. Curr. HIV Res. 2019, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Maundrell, K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 1993, 123, 127–130. [Google Scholar] [CrossRef]
- Zhao, Y.; Elder, R.T.; Chen, M.; Cao, J. Fission yeast expression vectors adapted for positive identification of gene insertion and green fluorescent protein fusion. Biotechniques 1998, 25, 438–440, 442, 444. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Elder, R.T.; Yu, M.; O’Gorman, M.G.; Selig, L.; Benarous, R.; Yamamoto, A.; Zhao, Y. Mutational analysis of Vpr-induced G2 arrest, nuclear localization, and cell death in fission yeast. J. Virol. 1999, 73, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Benko, Z.; Liang, D.; Agbottah, E.; Hou, J.; Chiu, K.; Yu, M.; Innis, S.; Reed, P.; Kabat, W.; Elder, R.T.; et al. Anti-Vpr activity of a yeast chaperone protein. J. Virol. 2004, 78, 11016–11029. [Google Scholar] [CrossRef][Green Version]
- Wire, M.B.; Shelton, M.J.; Studenberg, S. Fosamprenavir: Clinical pharmacokinetics and drug interactions of the amprenavir prodrug. Clin. Pharm. 2006, 45, 137–168. [Google Scholar] [CrossRef] [PubMed]
- Becker, Y. HIV-1 induced AIDS is an allergy and the allergen is the Shed gp120--a review, hypothesis, and implications. Virus Genes 2004, 28, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Furfine, E.S.; Baker, C.T.; Hale, M.R.; Reynolds, D.J.; Salisbury, J.A.; Searle, A.D.; Studenberg, S.D.; Todd, D.; Tung, R.D.; Spaltenstein, A. Preclinical pharmacology and pharmacokinetics of GW433908, a water-soluble prodrug of the human immunodeficiency virus protease inhibitor amprenavir. Antimicrob. Agents Chemother. 2004, 48, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Pas, T.; Bergonzi, A.; Lescrinier, E.; Vergauwen, B.; Van den Mooter, G. Drug-carrier binding and enzymatic carrier digestion in amorphous solid dispersions containing proteins as carrier. Int. J. Pharm. 2019, 563, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Griffin, L.; Annaert, P.; Brouwer, K.L. Influence of drug transport proteins on the pharmacokinetics and drug interactions of HIV protease inhibitors. J. Pharm. Sci. 2011, 100, 3636–3654. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.A.; German, P.; Murray, B.P.; Wei, L.; Jain, A.; West, S.; Warren, D.; Hui, J.; Kearney, B.P. Pharmacokinetics and pharmacodynamics of GS-9350: A novel pharmacokinetic enhancer without anti-HIV activity. Clin. Pharmacol. Ther. 2010, 87, 322–329. [Google Scholar] [CrossRef]
- McCoy, C.; Badowski, M.; Sherman, E.; Crutchley, R.; Smith, E.; Chastain, D.B.; Society of Infectious Diseases, P. Strength in amalgamation: Newer combination agents for HIV and implications for practice. Pharmacotherapy 2018, 38, 86–107. [Google Scholar] [CrossRef]
- Lamb, D.C.; Maspahy, S.; Kelly, D.E.; Manning, N.J.; Geber, A.; Bennett, J.E.; Kelly, S.L. Purification, reconstitution, and inhibition of cytochrome P-450 sterol delta22-desaturase from the pathogenic fungus Candida glabrata. Antimicrob. Agents Chemother. 1999, 43, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Durairaj, P.; Fan, L.; Du, W.; Ahmad, S.; Mebrahtu, D.; Sharma, S.; Ashraf, R.A.; Liu, J.; Liu, Q.; Bureik, M. Functional expression and activity screening of all human cytochrome P450 enzymes in fission yeast. FEBS Lett. 2019, 593, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, J.S.; Vermeulen, N.P.; Chris Vos, J. Yeast as a humanized model organism for biotransformation-related toxicity. Curr. Drug Metab. 2012, 13, 1464–1475. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, J.; Wan, D.F.; Gu, J.R.; Gong, Y.; Yang, S.L.; Hao, D.C.; Yang, L. Establishment of a yeast system that stably expresses human cytochrome P450 reductase: Application for the study of drug metabolism of cytochrome P450s in vitro. Protein Expr. Purif. 2006, 47, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Machalz, D.; Zollner, A.; Sorensen, E.J.; Wolber, G.; Bureik, M. Efficient substrate screening and inhibitor testing of human CYP4Z1 using permeabilized recombinant fission yeast. Biochem. Pharmacol. 2017, 146, 174–187. [Google Scholar] [CrossRef]
- Ali, A.; Reddy, G.S.; Nalam, M.N.; Anjum, S.G.; Cao, H.; Schiffer, C.A.; Rana, T.M. Structure-based design, synthesis, and structure-activity relationship studies of HIV-1 protease inhibitors incorporating phenyloxazolidinones. J. Med. Chem. 2010, 53, 7699–7708. [Google Scholar] [CrossRef][Green Version]
- Molina, J.M.; Cohen, C.; Katlama, C.; Grinsztejn, B.; Timerman, A.; Pedro Rde, J.; Vangeneugden, T.; Miralles, D.; Meyer, S.D.; Parys, W.; et al. Safety and efficacy of darunavir (TMC114) with low-dose ritonavir in treatment-experienced patients: 24-week results of POWER 3. JAIDS J. Acquir. Immune Defic. Syndr. 2007, 46, 24–31. [Google Scholar] [CrossRef]
- Zhao, Y.; Hopkins, K.M.; Lieberman, H.B. A method for the preparation and storage of frozen, competent Schizosaccharomyces pombe spheroplasts. Biotechniques 1993, 15, 238. [Google Scholar]
- Flor-Parra, I.; Zhurinsky, J.; Bernal, M.; Gallardo, P.; Daga, R.R. A Lallzyme MMX-based rapid method for fission yeast protoplast preparation. Yeast 2014, 31, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, T.; Giga-Hama, Y.; Takegawa, K. A survey of all 11 ABC transporters in fission yeast: Two novel ABC transporters are required for red pigment accumulation in a Schizosaccharomyces pombe adenine biosynthetic mutant. Microbiology 2006, 152, 2309–2321. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawashima, S.A.; Takemoto, A.; Nurse, P.; Kapoor, T.M. Analyzing fission yeast multidrug resistance mechanisms to develop a genetically tractable model system for chemical biology. Chem. Biol. 2012, 19, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Aoi, Y.; Sato, M.; Sutani, T.; Shirahige, K.; Kapoor, T.M.; Kawashima, S.A. Dissecting the first and the second meiotic divisions using a marker-less drug-hypersensitive fission yeast. Cell Cycle 2014, 13, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Kurisawa, N.; Yukawa, M.; Koshino, H.; Onodera, T.; Toda, T.; Kimura, K.I. Kolavenic acid analog restores growth in HSET-overproducing fission yeast cells and multipolar mitosis in MDA-MB-231 human cells. Bioorg. Med. Chem. 2020, 28, 115154. [Google Scholar] [CrossRef] [PubMed]
- Krohn, A.; Redshaw, S.; Ritchie, J.C.; Graves, B.J.; Hatada, M.H. Novel binding mode of highly potent HIV-proteinase inhibitors incorporating the (R)-hydroxyethylamine isostere. J. Med. Chem. 1991, 34, 3340–3342. [Google Scholar] [CrossRef]
- Requena, D.G.D.; Gallego, O.; Mendoza, C.D.; Corral, A.; Jimenez-Nacher, I.; Soriano, V. Indinavir plasma concentrations and resistance mutations in patients experiencing early virological failure. AIDS Res. Hum. Retrovir. 2003, 19, 457–459. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. Hiv/aids 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Sham, H.L.; Kempf, D.J.; Molla, A.; Marsh, K.C.; Kumar, G.N.; Chen, C.M.; Kati, W.; Stewart, K.; Lal, R.; Hsu, A.; et al. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents Chemother. 1998, 42, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Becker, S. Atazanavir: Improving the HIV protease inhibitor class. Expert Rev. Anti Infect. Ther. 2003, 1, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Larder, B.A.; Hertogs, K.; Bloor, S.; Van den Eynde, C.H.; DeCian, W.; Wang, Y.; Freimuth, W.W.; Tarpley, G. Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples. AIDS 2000, 14, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, R.S.; Goa, K.L. Lopinavir/ritonavir: A review of its use in the management of HIV infection. Drugs 2003, 63, 769–802. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.M.; Schafer, J.J.; Priano, J.; Squires, K.E. Cobicistat: A new boost for the treatment of human immunodeficiency virus infection. Pharmacotherapy 2013, 33, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.A.; Fernandes, P.A.; Ramos, M.J. Substrate recognition in HIV-1 protease: A computational study. J. Phys. Chem. B 2010, 114, 2525–2532. [Google Scholar] [CrossRef]
- Benko, Z.; Elder, R.T.; Liang, D.; Zhao, R.Y. Fission yeast as a HTS platform for molecular probes of HIV-1 Vpr-induced cell death. Int. J. High Throughput Screen 2010, 1, 151–162. [Google Scholar]
- Zhao, R.Y. Yeast for virus research. Microb. Cell 2017, 4, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lieberman, H.B. Schizosaccharomyces pombe: A model for molecular studies of eukaryotic genes. DNA Cell Biol. 1995, 14, 359–371. [Google Scholar] [CrossRef]
- Li, G.; Zhao, R.Y. Molecular cloning and characterization of small viral genome in fission yeast. Methods Mol. Biol. 2018, 1721, 47–61. [Google Scholar] [CrossRef]
- Agniswamy, J.; Shen, C.H.; Wang, Y.F.; Ghosh, A.K.; Rao, K.V.; Xu, C.X.; Sayer, J.M.; Louis, J.M.; Weber, I.T. Extreme multidrug resistant HIV-1 protease with 20 mutations is resistant to novel protease inhibitors with P1′-pyrrolidinone or P2-tris-tetrahydrofuran. J. Med. Chem. 2013, 56, 4017–4027. [Google Scholar] [CrossRef]
- Koh, Y.; Amano, M.; Towata, T.; Danish, M.; Leshchenko-Yashchuk, S.; Das, D.; Nakayama, M.; Tojo, Y.; Ghosh, A.K.; Mitsuya, H. In vitro selection of highly darunavir-resistant and replication-competent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors. J. Virol. 2010, 84, 11961–11969. [Google Scholar] [CrossRef] [PubMed]
- Chetty, S.; Bhakat, S.; Martin, A.J.; Soliman, M.E. Multi-drug resistance profile of PR20 HIV-1 protease is attributed to distorted conformational and drug binding landscape: Molecular dynamics insights. J. Biomol. Struct. Dyn. 2016, 34, 135–151. [Google Scholar] [CrossRef]
- Elder, R.T.; Yu, M.; Chen, M.; Edelson, S.; Zhao, Y. Cell cycle G2 arrest induced by HIV-1 Vpr in fission yeast (Schizosaccharomyces pombe) is independent of cell death and early genes in the DNA damage checkpoint. Viral Res. 2000, 68, 161–173. [Google Scholar] [CrossRef]
- Benko, Z.; Zhao, R.Y. Zeocin for selection of bleMX6 resistance in fission yeast. Biotechniques 2011, 51, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Basi, G.; Schmid, E.; Maundrell, K. TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene 1993, 123, 131–136. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, J.; O’Gorman, M.R.; Yu, M.; Yogev, R. Effect of human immunodeficiency virus type 1 protein R (vpr) gene expression on basic cellular function of fission yeast Schizosaccharomyces pombe. J. Virol. 1996, 70, 5821–5826. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Abbreviation | Brand Name | Molecular Weight (g/mol) ** | Drug Manufacturer | Pure Compound Source ‡ (ARP#) |
|---|---|---|---|---|---|
| First-generation single-agent drug | |||||
| Saquinavir | SQV | Invirase | 670.84 | F. Hoffmann-La Roche | 4658 |
| Indinavir | IDV | Crixivan | 613.79 | Merck | 8145 |
| Ritonavir | RTV | Norvir | 720.94 | Abbott Labs | 4622 |
| Nelfinavir | NFV | Viracept | 567.78 | Pfizer | 4621 |
| Amprenavir | APV | Agenerase | 505.63 | Vertex Pharmaceuticals | 8148 |
| Fosamprenavir * | FPV | Lexiva | 585.61 | GlaxoSmithKine | na |
| Lopinavir | LPV | Kaletra | 628.81 | Abbott Labs | 9481 |
| Second-generation single-agent drug | |||||
| Atazanavir | ATV | Reyataz | 704.86 | Bristol-Myers Squibb | 10,003 |
| Tipranavir | TPV | Aptivus | 602.66 | Boehringer Ingelheim | 11,285 |
| Darunavir | DRV | Prezista | 547.66 | Tibotec | 11,447 |
| Fixed dose combination drug | |||||
| Lopinavir /Ritonavir | LPV/r | Kaletra | 628.81 /720.94 | Abbott Labs | na |
| Atazanavir/ Cobicistat | ATV/co | Evotaz | 704.86 | Bristol-Myers Squibb | na |
| Darunavir/ Cobicistat | DRV/co | Prezcobix | 547.66 | Tibotec | na |
| HIV-1 Protease Inhibitor Drug | EC50 in Human Cell (nM) 1 | EC50 in Fission Yeast /Pure Compound (μM) 2 | EC50 in Fission Yeast /Drug Formulation (μM) 3 | Reference |
|---|---|---|---|---|
| First-generation single-agent drug | ||||
| Saquinavir (SQV) | 5–25 | 9.88 ± 0.25 | nd | [61] |
| Indinavir (IDV) | 5–30 | 48.64 ± 2.51 | nd | [62] |
| Ritonavir (RTV) | 40–100 | 115.2 ± 3.2 | 70.39 ± 2.25 | [53,63] |
| Nelfinavir (NFV) | 3–40 | 9.7 ± 0.34 | nd | [53,63] |
| Amprenavir (APV) | 15–60 | 55.41 ± 3.18 | nd | [18] |
| Fosamprenavir (FPV) * | cnd | na | cnd | [18] |
| Lopinavir (LPV) | 4–11 | 4.59 ± 0.06 | nd | [64] |
| Second-generation single-agent drug | ||||
| Atazanavir (ATV) | 2–5 | 21.08 ± 0.07 | 16.33 ± 0.95 | [65] |
| Tipranavir (TPV) | 30–70 | 7.86 ± 0.32 | nd | [66] |
| Darunavir (DRV) | 1–10 | 2.53 ± 0.08 | 1.81 ± 0.08 | [54] |
| Fixed dose combination drug | ||||
| Lopinavir/Ritonavir (LPV/r) | na | na | 4.03 ± 0.74 | [67] |
| Atazanavir/ Cobicistat (ATV/co) | na | na | 17.04 ± 0.95 | [68] |
| Darunavir/Cobicistat (DRV/co) | 1–2 | na | 2.02 ± 0.15 * | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Vernon, K.; Li, Q.; Benko, Z.; Amoroso, A.; Nasr, M.; Zhao, R.Y. Single-Agent and Fixed-Dose Combination HIV-1 Protease Inhibitor Drugs in Fission Yeast (Schizosaccharomyces pombe). Pathogens 2021, 10, 804. https://doi.org/10.3390/pathogens10070804
Zhang J, Vernon K, Li Q, Benko Z, Amoroso A, Nasr M, Zhao RY. Single-Agent and Fixed-Dose Combination HIV-1 Protease Inhibitor Drugs in Fission Yeast (Schizosaccharomyces pombe). Pathogens. 2021; 10(7):804. https://doi.org/10.3390/pathogens10070804
Chicago/Turabian StyleZhang, Jiantao, Kasey Vernon, Qi Li, Zsigmond Benko, Anthony Amoroso, Mohamed Nasr, and Richard Y. Zhao. 2021. "Single-Agent and Fixed-Dose Combination HIV-1 Protease Inhibitor Drugs in Fission Yeast (Schizosaccharomyces pombe)" Pathogens 10, no. 7: 804. https://doi.org/10.3390/pathogens10070804
APA StyleZhang, J., Vernon, K., Li, Q., Benko, Z., Amoroso, A., Nasr, M., & Zhao, R. Y. (2021). Single-Agent and Fixed-Dose Combination HIV-1 Protease Inhibitor Drugs in Fission Yeast (Schizosaccharomyces pombe). Pathogens, 10(7), 804. https://doi.org/10.3390/pathogens10070804