
Genetic Variation in the Glycoprotein B Sequence of Equid Herpesvirus 5 among Horses of Various Breeds at Polish National Studs




Abstract
1. Introduction
2. Results
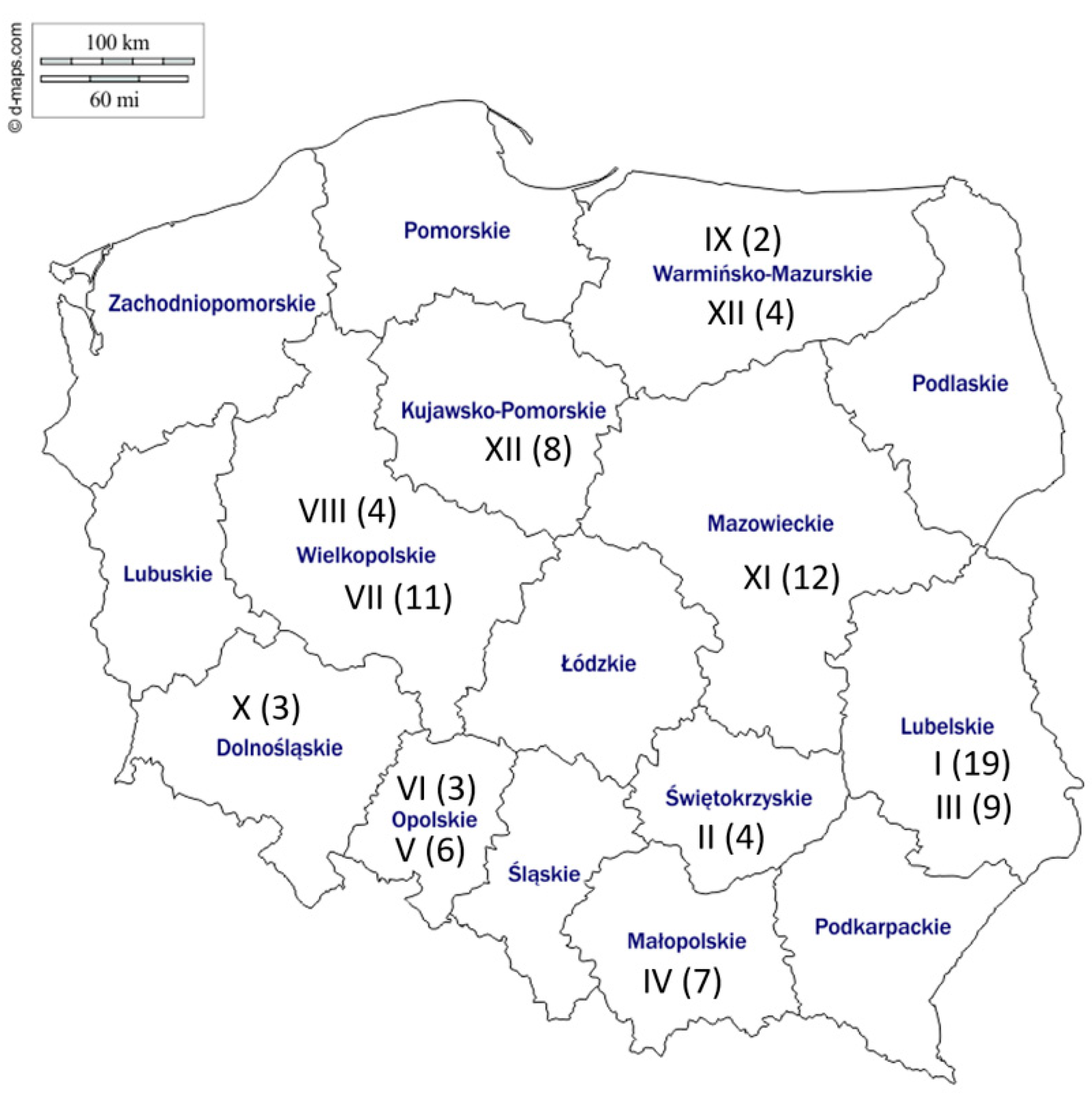
2.1. Sequence Analysis
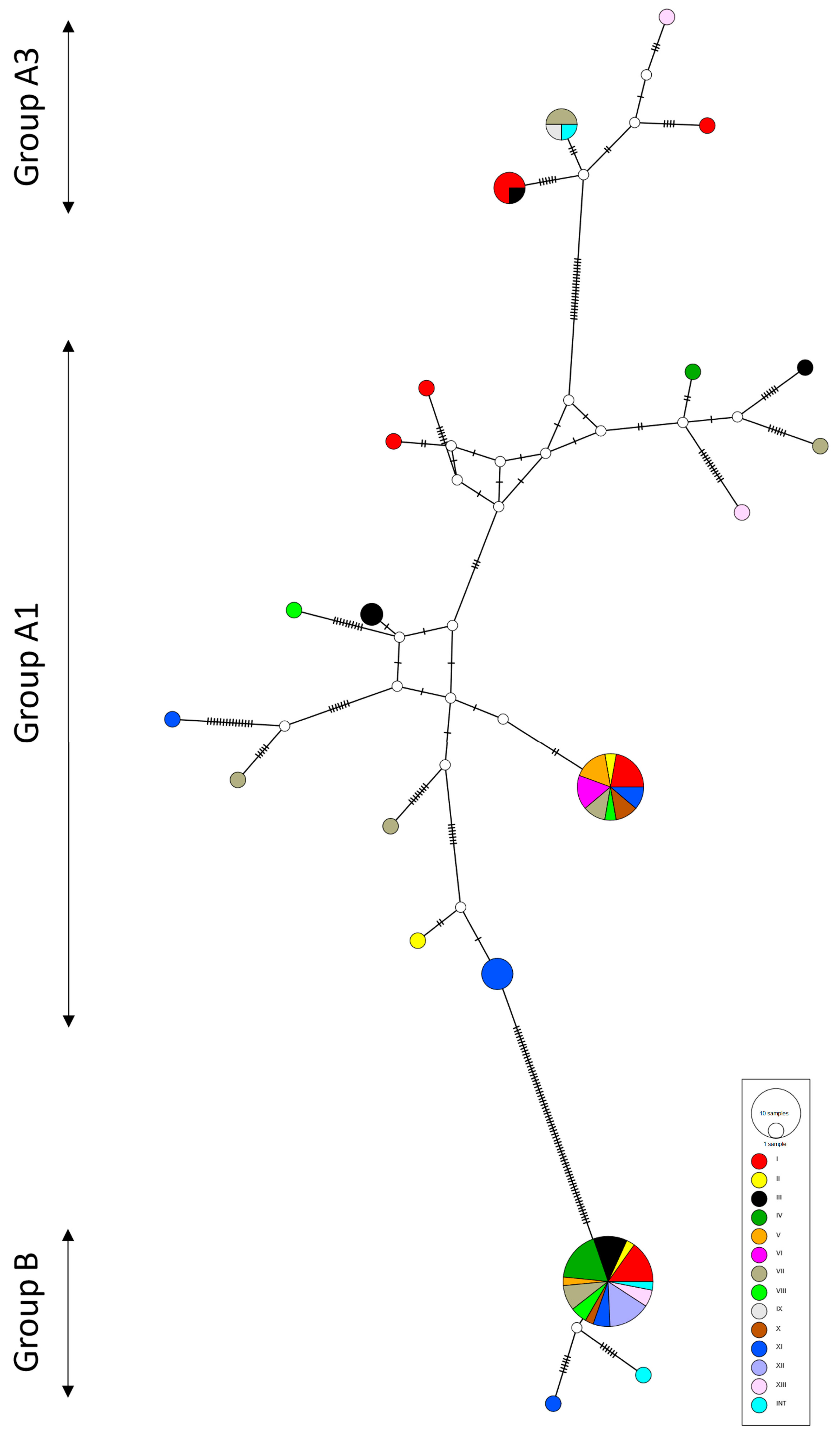
2.2. Phylogeny
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Conventional PCR and Sequencing
4.3. Sequence Analyses
4.4. GenBank Accession Numbers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marenzoni, M.L.; Stefanetti, V.; Danzetta, M.L.; Timoney, P.J. Gammaherpesvirus infections in equids: A review. Vet. Med. Auckl. 2015, 6, 91–101. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Telford, E.A.; Studdert, M.J.; Agius, C.T.; Watson, M.S.; Aird, H.C.; Davison, A.J. Equine herpesviruses 2 and 5 are gamma-herpesviruses. Virology 1993, 195, 492–499. [Google Scholar] [CrossRef]
- Marenzoni, M.L.; Coppola, G.; Maranesi, M.; Passamonti, F.; Cappelli, K.; Capomaccio, S.; Verini Supplizi, A.; Thiry, E.; Coletti, M. Age-dependent prevalence of equid herpesvirus 5 infection. Vet. Res. Commun. 2010, 34, 703–708. [Google Scholar] [CrossRef]
- Wang, L.; Raidal, S.L.; Pizzirani, A.; Wilcox, G.E. Detection of respiratory herpesviruses in foals and adult horses determined by nested multiplex PCR. Vet. Microbiol. 2007, 121, 18–28. [Google Scholar] [CrossRef]
- Hue, E.S.; Fortier, G.D.; Fortier, C.I.; Leon, A.M.; Richard, E.A.; Legrand, L.J.; Pronost, S.L. Detection and quantitation of equid gammaherpesviruses (EHV-2, EHV-5) in nasal swabs using an accredited standardised quantitative PCR method. J. Virol. Methods 2014, 198, 18–25. [Google Scholar] [CrossRef]
- Mekuria, Z.H.; El-Hage, C.; Ficorilli, N.P.; Washington, E.A.; Gilkerson, J.R.; Hartley, C.A. Mapping B lymphocytes as major reservoirs of naturally occurring latent equine herpesvirus 5 infection. J. Gen. Virol 2017, 98, 461–470. [Google Scholar] [CrossRef]
- Ackermann, M. Pathogenesis of gammaherpesvirus infections. Vet. Microbiol. 2006, 113, 211–222. [Google Scholar] [CrossRef]
- Hartley, C.A.; Dynon, K.J.; Mekuria, Z.H.; El-Hage, C.M.; Holloway, S.A.; Gilkerson, J.R. Equine gammaherpesviruses: Perfect parasites? Vet. Microbiol. 2013, 167, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J. Gammaherpesviruses and pulmonary fibrosis: Evidence from humans, horses, and rodents. Vet. Pathol. 2014, 51, 372–384. [Google Scholar] [CrossRef]
- Williams, K.J.; Robinson, N.E.; Lim, A.; Brandenberger, C.; Maes, R.; Behan, A.; Bolin, S.R. Experimental induction of pulmonary fibrosis in horses with the gammaherpesvirus equine herpesvirus 5. PLoS ONE 2013, 8, e77754. [Google Scholar] [CrossRef]
- Wilkie, G.S.; Kerr, K.; Stewart, J.P.; Studdert, M.J.; Davison, A.J. Genome sequences of equid herpesviruses 2 and 5. Genome Announc. 2015, 3. [Google Scholar] [CrossRef]
- Agius, C.T.; Nagesha, H.S.; Studdert, M.J. Equine herpesvirus 5: Comparisons with EHV2 (equine cytomegalovirus), cloning, and mapping of a new equine herpesvirus with a novel genome structure. Virology 1992, 191, 176–186. [Google Scholar] [CrossRef]
- Pereira, L. Function of glycoprotein B homologues of the family herpesviridae. Infect. Agents Dis. 1994, 3, 9–28. [Google Scholar]
- Sathiyamoorthy, K.; Chen, J.; Longnecker, R.; Jardetzky, T.S. The COMPLEXity in herpesvirus entry. Curr. Opin. Virol. 2017, 24, 97–104. [Google Scholar] [CrossRef]
- Holloway, S.A.; Lindquester, G.J.; Studdert, M.J.; Drummer, H.E. Identification, sequence analysis and characterisation of equine herpesvirus 5 glycoprotein B. Arch. Virol. 1999, 144, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Holloway, S.A.; Studdert, M.J.; Drummer, H.E. Characterization of glycoprotein B of the gammaherpesvirus equine herpesvirus-2. J. Gen. Virol. 1998, 79, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Mobeen, F.; Prakash, T. Comparative Genomics of Herpesviridae Family to Look for Potential Signatures of Human Infecting Strains. Int. J. Genomics 2016, 2016, 9543274. [Google Scholar] [CrossRef]
- Stasiak, K.; Dunowska, M.; Rola, J. Prevalence and sequence analysis of equid herpesviruses from the respiratory tract of Polish horses. Virol. J. 2018, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Holloway, S.A.; Lindquester, G.J.; Studdert, M.J.; Drummer, H.E. Analysis of equine herpesvirus 2 strain variation using monoclonal antibodies to glyucoprotein B. Arch. Virol. 2000, 145, 1699–1713. [Google Scholar] [CrossRef]
- Brault, S.A.; Bird, B.H.; Balasuriya, U.B.; MacLachlan, N.J. Genetic heterogeneity and variation in viral load during equid herpesvirus-2 infection of foals. Vet. Microbiol 2011, 147, 253–261. [Google Scholar] [CrossRef]
- Bell, S.A.; Balasuriya, U.B.; Gardner, I.A.; Barry, P.A.; Wilson, W.D.; Ferraro, G.L.; MacLachlan, N.J. Temporal detection of equine herpesvirus infections of a cohort of mares and their foals. Vet. Microbiol. 2006, 116, 249–257. [Google Scholar] [CrossRef]
- Lee, S.K.; Lee, I. The Molecular Detection of Equine Herpesviruses 2 and 5 in Genital Swabs From Clinically Normal Thoroughbred Mares in South Korea. J. Equine Vet. Sci. 2019, 79, 68–72. [Google Scholar] [CrossRef]
- Negussie, H.; Gizaw, D.; Tesfaw, L.; Li, Y.; Oguma, K.; Sentsui, H.; Tessema, T.S.; Nauwynck, H.J. Detection of Equine Herpesvirus (EHV) -1, -2, -4 and -5 in Ethiopian Equids with and without Respiratory Problems and Genetic Characterization of EHV-2 and EHV-5 Strains. Transbound Emerg. Dis. 2017, 64, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Ataseven, V.S.; Bilge-Dagalp, S.; Oguzoglu, T.C.; Karapinar, Z.; Guzel, M.; Tan, M.T. Detection and sequence analysis of equine gammaherpesviruses from horses with respiratory tract disease in Turkey. Transbound Emerg. Dis. 2010, 57, 271–276. [Google Scholar] [CrossRef]
- Radalj, A.; Nišavić, J.; Krnjaić, D.; Valčić, M.; Jovanović, T.; Veljović, L.; Milić, N. Detection and molecular characterization of equine herpesviruses 1, 2, and 5 in horses in the Republic of Serbia. Acta Vet. Brno 2018, 87, 27–34. [Google Scholar] [CrossRef]
- Dunowska, M.; Holloway, S.A.; Wilks, C.R.; Meers, J. Genomic variability of equine herpesvirus-5. Arch. Virol. 2000, 145, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Back, H.; Ullman, K.; Leijon, M.; Soderlund, R.; Penell, J.; Stahl, K.; Pringle, J.; Valarcher, J.F. Genetic variation and dynamics of infections of equid herpesvirus 5 in individual horses. J. Gen. Virol. 2016, 97, 169–178. [Google Scholar] [CrossRef]
- Torfason, E.G.; Thorsteinsdottir, L.; Torsteinsdottir, S.; Svansson, V. Study of equid herpesviruses 2 and 5 in Iceland with a type-specific polymerase chain reaction. Res. Vet. Sci. 2008, 85, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.G.; Horst, J.A.; Rose, T.M. Conservation of the glycoprotein B homologs of the Kaposis sarcoma-associated herpesvirus (KSHV/HHV8) and old world primate rhadinoviruses of chimpanzees and macaques. Virology 2016, 494, 29–46. [Google Scholar] [CrossRef]
- Coaquette, A.; Bourgeois, A.; Dirand, C.; Varin, A.; Chen, W.; Herbein, G. Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin. Infect. Dis. 2004, 39, 155–161. [Google Scholar] [CrossRef]
- Norberg, P.; Depledge, D.P.; Kundu, S.; Atkinson, C.; Brown, J.; Haque, T.; Hussaini, Y.; MacMahon, E.; Molyneaux, P.; Papaevangelou, V.; et al. Recombination of Globally Circulating Varicella-Zoster Virus. J. Virol. 2015, 89, 7133–7146. [Google Scholar] [CrossRef]
- Fakhri, O.; Devlin, J.M.; Browning, G.F.; Vaz, P.K.; Thilakarathne, D.; Lee, S.W.; Hartley, C.A. Genomic recombination between infectious laryngotracheitis vaccine strains occurs under a broad range of infection conditions in vitro and in ovo. PLoS ONE 2020, 15, e0229082. [Google Scholar] [CrossRef]
- Dewals, B.; Thirion, M.; Markine-Goriaynoff, N.; Gillet, L.; de Fays, K.; Minner, F.; Daix, V.; Sharp, P.M.; Vanderplasschen, A. Evolution of Bovine herpesvirus 4: Recombination and transmission between African buffalo and cattle. J. Gen. Virol. 2006, 87, 1509–1519. [Google Scholar] [CrossRef]
- Tian, S. A 20 Residues Motif Delineates the Furin Cleavage Site and its Physical Properties May Influence Viral Fusion. Biochem. Insights 2009, 2, 9–20. [Google Scholar] [CrossRef]
- Braun, E.; Sauter, D. Furin-mediated protein processing in infectious diseases and cancer. Clin. Transl. Immunol. 2019, 8, e1073. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Su, S.; Wang, X.; Xu, W.; Liu, Z.; Zhu, Y.; Wang, Q.; Lu, L.; Jiang, S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal. Transduct Target. Ther. 2020, 5, 92. [Google Scholar] [CrossRef]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef]
- Bolt, G.; Pedersen, L.O.; Birkeslund, H.H. Cleavage of the respiratory syncytial virus fusion protein is required for its surface expression: Role of furin. Virus Res. 2000, 68, 25–33. [Google Scholar] [CrossRef]
- Mozzi, A.; Biolatti, M.; Cagliani, R.; Forni, D.; Dell’Oste, V.; Pontremoli, C.; Vantaggiato, C.; Pozzoli, U.; Clerici, M.; Landolfo, S.; et al. Past and ongoing adaptation of human cytomegalovirus to its host. PLoS Pathog. 2020, 16, e1008476. [Google Scholar] [CrossRef]
- Strive, T.; Borst, E.; Messerle, M.; Radsak, K. Proteolytic processing of human cytomegalovirus glycoprotein B is dispensable for viral growth in culture. J. Virol. 2002, 76, 1252–1264. [Google Scholar] [CrossRef]
- Norais, N.; Tang, D.; Kaur, S.; Chamberlain, S.H.; Masiarz, F.R.; Burke, R.L.; Marcus, F. Disulfide bonds of herpes simplex virus type 2 glycoprotein gB. J. Virol. 1996, 70, 7379–7387. [Google Scholar] [CrossRef]
- Lang, S.M.; Means, R.E. Characterization of cytoplasmic motifs important in rhesus rhadinovirus gB processing and trafficking. Virology 2010, 398, 233–242. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Leigh, J.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Variation within Populations | Variation between Populations | PhiPT | P |
|---|---|---|---|---|
| Geographical location (studs I–XIII, INT) | 93% | 7% | 0.07 | 0.15 |
| Age (<1, 1, 2–5, 6–10, 11–15, >15 years, UNK) | 93% | 7% | 0.06 | 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stasiak, K.; Dunowska, M.; Trewick, S.; Rola, J. Genetic Variation in the Glycoprotein B Sequence of Equid Herpesvirus 5 among Horses of Various Breeds at Polish National Studs. Pathogens 2021, 10, 322. https://doi.org/10.3390/pathogens10030322
Stasiak K, Dunowska M, Trewick S, Rola J. Genetic Variation in the Glycoprotein B Sequence of Equid Herpesvirus 5 among Horses of Various Breeds at Polish National Studs. Pathogens. 2021; 10(3):322. https://doi.org/10.3390/pathogens10030322
Chicago/Turabian StyleStasiak, Karol, Magdalena Dunowska, Steven Trewick, and Jerzy Rola. 2021. "Genetic Variation in the Glycoprotein B Sequence of Equid Herpesvirus 5 among Horses of Various Breeds at Polish National Studs" Pathogens 10, no. 3: 322. https://doi.org/10.3390/pathogens10030322
APA StyleStasiak, K., Dunowska, M., Trewick, S., & Rola, J. (2021). Genetic Variation in the Glycoprotein B Sequence of Equid Herpesvirus 5 among Horses of Various Breeds at Polish National Studs. Pathogens, 10(3), 322. https://doi.org/10.3390/pathogens10030322