
Bacteria and Host Interplay in Staphylococcus aureus Septic Arthritis and Sepsis


Abstract
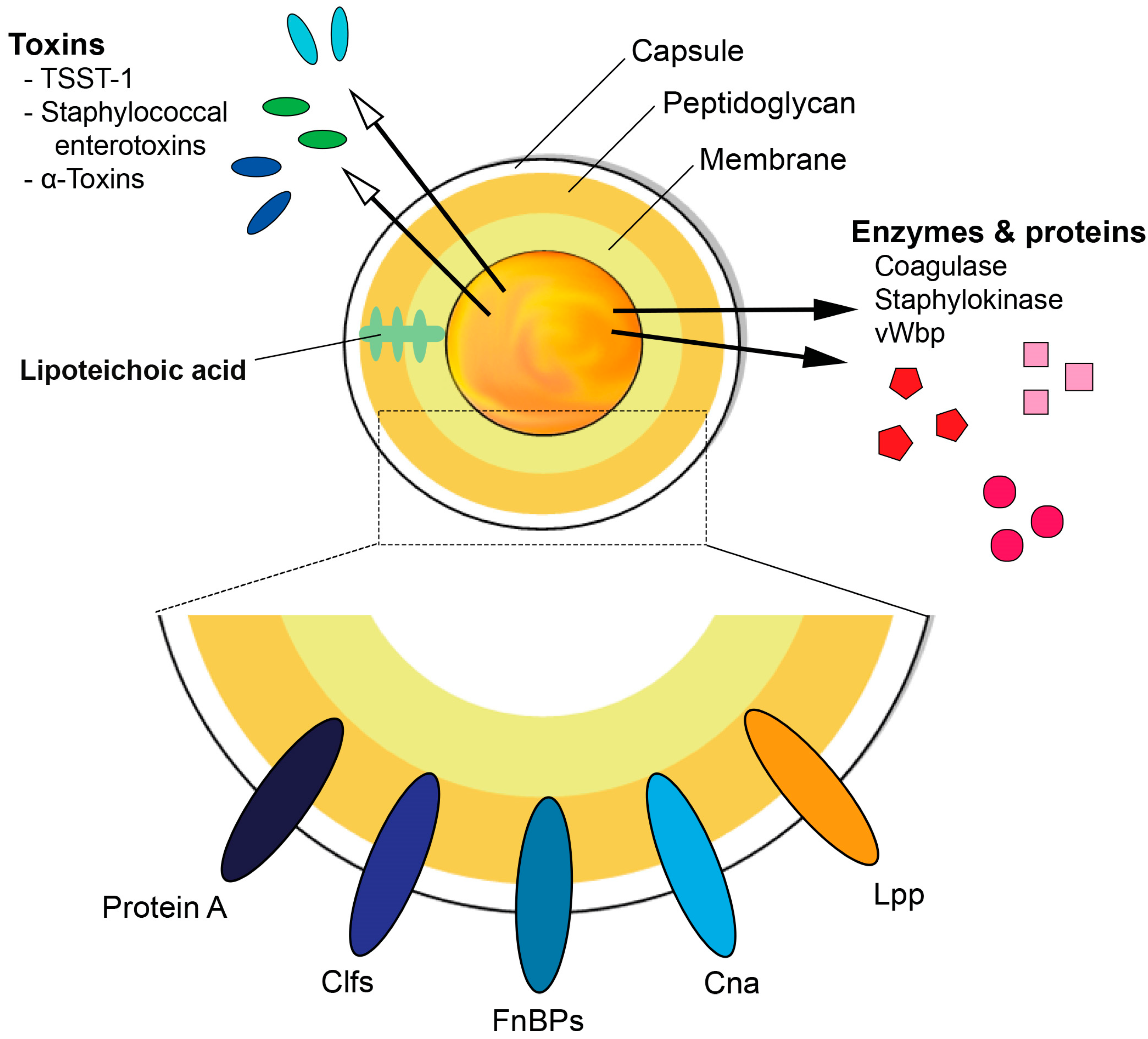
1. S. aureus and Its Virulence Factors
1.1. Cell Wall Components
1.2. Surface Proteins
1.3. Secreted Proteins
1.4. Toxins
1.5. Bacterial DNA
2. The Immune Response during S. aureus Infections
2.1. Innate Immunity
2.2. Neutrophils
2.3. Macrophages
2.4. Natural Killer (NK) Cells
2.5. The Complement System
2.6. Adaptive Immunity
2.6.1. T-Cells
2.6.2. Natural Killer T (NKT) Cells
2.6.3. B-Cells
2.6.4. Other Cell Types
2.6.5. Cytokines
3. Septic Arthritis and Sepsis
4. Bacteria and Host Interplay Determines the Disease Outcome
4.1. Joint Affinity of S. aureus is the Key Mechanism of Septic Arthritis
4.2. The Impact of Bacteria and Host Interplay on Biofilm Formation in Septic Arthritis
4.3. The Role of Microbiome in Septic Arthritis and Sepsis
4.4. Exaggerated Immune Response Causes Joint Damage in Septic Arthritis
4.5. Cytokine Storm and Immune Paralysis in S. aureus Sepsis
5. Therapies Targeting Host and Bacteria Interaction
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wertheim, H.F.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Bien, J.; Sokolova, O.; Bozko, P. Characterization of Virulence Factors of Staphylococcus aureus: Novel Function of Known Virulence Factors That Are Implicated in Activation of Airway Epithelial Proinflammatory Response. J. Pathog. 2011, 2011, 601905. [Google Scholar] [CrossRef] [PubMed]
- Ali, A. Biologics in Staphylococcus Aureus Arthritis. Ph.D. Thesis, University of Gothenburg, Gothenburg, Sweden, 2016. [Google Scholar]
- Nanra, J.S.; Buitrago, S.M.; Crawford, S.; Ng, J.; Fink, P.S.; Hawkins, J.; Scully, I.L.; McNeil, L.K.; Aste-Amezaga, J.M.; Cooper, D.; et al. Capsular polysaccharides are an important immune evasion mechanism for Staphylococcus aureus. Hum. Vaccin. Immunother. 2013, 9, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.M.; Lee, J.C.; Bremell, T.; Ryden, C.; Tarkowski, A. The role of staphylococcal polysaccharide microcapsule expression in septicemia and septic arthritis. Infect. Immun. 1997, 65, 4216–4221. [Google Scholar] [CrossRef]
- Watts, A.; Ke, D.; Wang, Q.; Pillay, A.; Nicholson-Weller, A.; Lee, J.C. Staphylococcus aureus strains that express serotype 5 or serotype 8 capsular polysaccharides differ in virulence. Infect. Immun. 2005, 73, 3502–3511. [Google Scholar] [CrossRef]
- Sharif, S.; Singh, M.; Kim, S.J.; Schaefer, J. Staphylococcus aureus peptidoglycan tertiary structure from carbon-13 spin diffusion. J. Am. Chem. Soc. 2009, 131, 7023–7030. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Willey, J.M.; Sherwood, L.; Woolverton, C.J. Prescott’s Microbiology, 9th ed.; McGraw-Hill: New York, NY, USA, 2014; p. 1. [Google Scholar]
- Grundling, A.; Schneewind, O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J. Bacteriol. 2006, 188, 2463–2472. [Google Scholar] [CrossRef]
- Kashyap, D.R.; Wang, M.; Liu, L.H.; Boons, G.J.; Gupta, D.; Dziarski, R. Peptidoglycan recognition proteins kill bacteria by activating protein-sensing two-component systems. Nat. Med. 2011, 17, 676–683. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Deng, G.M.; Foster, S.; Tarkowski, A. Staphylococcal peptidoglycans induce arthritis. Arthritis Res. Ther. 2001, 3, 375–380. [Google Scholar] [CrossRef]
- Dusad, A.; Thiele, G.M.; Klassen, L.W.; Gleason, A.M.; Bauer, C.; Mikuls, T.R.; Duryee, M.J.; West, W.W.; Romberger, D.J.; Poole, J.A. Organic dust, lipopolysaccharide, and peptidoglycan inhalant exposures result in bone loss/disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.; Nguyen, M.T.; Engdahl, C.; Na, M.; Jarneborn, A.; Hu, Z.; Karlsson, A.; Pullerits, R.; Ali, A.; Gotz, F.; et al. The YIN and YANG of lipoproteins in developing and preventing infectious arthritis by Staphylococcus aureus. PLoS Pathog. 2019, 15, e1007877. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Gotz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Sychantha, D.; Jones, C.S.; Little, D.J.; Moynihan, P.J.; Robinson, H.; Galley, N.F.; Roper, D.I.; Dowson, C.G.; Howell, P.L.; Clarke, A.J. In vitro characterization of the antivirulence target of Gram-positive pathogens, peptidoglycan O-acetyltransferase A (OatA). PLoS Pathog. 2017, 13, e1006667. [Google Scholar] [CrossRef] [PubMed]
- Baranwal, G.; Mohammad, M.; Jarneborn, A.; Reddy, B.R.; Golla, A.; Chakravarty, S.; Biswas, L.; Gotz, F.; Shankarappa, S.; Jin, T.; et al. Impact of cell wall peptidoglycan O-acetylation on the pathogenesis of Staphylococcus aureus in septic arthritis. Int. J. Med. Microbiol. 2017, 307, 388–397. [Google Scholar] [CrossRef]
- Reichmann, N.T.; Grundling, A. Location, synthesis and function of glycolipids and polyglycerolphosphate lipoteichoic acid in Gram-positive bacteria of the phylum Firmicutes. FEMS Microbiol. Lett. 2011, 319, 97–105. [Google Scholar] [CrossRef]
- Brown, S.; Santa Maria, J.P., Jr.; Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef]
- Morath, S.; Geyer, A.; Hartung, T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J. Exp. Med. 2001, 193, 393–397. [Google Scholar] [CrossRef]
- Morath, S.; Stadelmaier, A.; Geyer, A.; Schmidt, R.R.; Hartung, T. Synthetic lipoteichoic acid from Staphylococcus aureus is a potent stimulus of cytokine release. J. Exp. Med. 2002, 195, 1635–1640. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Gotz, F. Lipoproteins of Gram-Positive Bacteria: Key Players in the Immune Response and Virulence. Microbiol. Mol. Biol. Rev. 2016, 80, 891–903. [Google Scholar] [CrossRef]
- Sheldon, J.R.; Heinrichs, D.E. The iron-regulated staphylococcal lipoproteins. Front. Cell. Infect. Microbiol. 2012, 2, 41. [Google Scholar] [CrossRef] [PubMed]
- Hammer, N.D.; Skaar, E.P. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 2011, 65, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Uebele, J.; Kumari, N.; Nakayama, H.; Peter, L.; Ticha, O.; Woischnig, A.K.; Schmaler, M.; Khanna, N.; Dohmae, N.; et al. Lipid moieties on lipoproteins of commensal and non-commensal staphylococci induce differential immune responses. Nat. Commun. 2017, 8, 2246. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.; Hu, Z.; Ali, A.; Kopparapu, P.K.; Na, M.; Jarneborn, A.; Stroparo, M.d.N.; Nguyen, M.-T.; Karlsson, A.; Götz, F.; et al. The role of Staphylococcus aureus lipoproteins in hematogenous septic arthritis. Sci. Rep. 2020, 10, 7936. [Google Scholar] [CrossRef]
- Schmaler, M.; Jann, N.J.; Ferracin, F.; Landolt, L.Z.; Biswas, L.; Gotz, F.; Landmann, R. Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J. Immunol. 2009, 182, 7110–7118. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Kraft, B.; Yu, W.; Demircioglu, D.D.; Hertlein, T.; Burian, M.; Schmaler, M.; Boller, K.; Bekeredjian-Ding, I.; Ohlsen, K.; et al. The νSaα Specific Lipoprotein Like Cluster (lpl) of S. aureus USA300 Contributes to Immune Stimulation and Invasion in Human Cells. PLoS Pathog. 2015, 11, e1004984. [Google Scholar] [CrossRef]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Hook, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef]
- Hair, P.S.; Echague, C.G.; Sholl, A.M.; Watkins, J.A.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect. Immun. 2010, 78, 1717–1727. [Google Scholar] [CrossRef]
- Josefsson, E.; Hartford, O.; O’Brien, L.; Patti, J.M.; Foster, T. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J. Infect. Dis. 2001, 184, 1572–1580. [Google Scholar] [CrossRef][Green Version]
- Kobayashi, S.D.; DeLeo, F.R. Staphylococcus aureus protein A promotes immune suppression. MBio 2013, 4, e00764-13. [Google Scholar] [CrossRef]
- Palmqvist, N.; Foster, T.; Tarkowski, A.; Josefsson, E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb. Pathog. 2002, 33, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, N.; Foster, T.; Fitzgerald, J.R.; Josefsson, E.; Tarkowski, A. Fibronectin-binding proteins and fibrinogen-binding clumping factors play distinct roles in staphylococcal arthritis and systemic inflammation. J. Infect. Dis. 2005, 191, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Switalski, L.M.; Patti, J.M.; Butcher, W.; Gristina, A.G.; Speziale, P.; Hook, M. A collagen receptor on Staphylococcus aureus strains isolated from patients with septic arthritis mediates adhesion to cartilage. Mol. Microbiol. 1993, 7, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Patti, J.M.; Bremell, T.; Krajewska-Pietrasik, D.; Abdelnour, A.; Tarkowski, A.; Ryden, C.; Hook, M. The Staphylococcus aureus collagen adhesin is a virulence determinant in experimental septic arthritis. Infect. Immun. 1994, 62, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Vanassche, T.; Verhaegen, J.; Peetermans, W.E.; van Ryn, J.; Cheng, A.; Schneewind, O.; Hoylaerts, M.F.; Verhamme, P. Inhibition of staphylothrombin by dabigatran reduces Staphylococcus aureus virulence. J. Thromb. Haemost. 2011, 9, 2436–2446. [Google Scholar] [CrossRef]
- Na, M.; Hu, Z.; Mohammad, M.; Stroparo, M.D.N.; Ali, A.; Fei, Y.; Jarneborn, A.; Verhamme, P.; Schneewind, O.; Missiakas, D.; et al. The Expression of von Willebrand Factor-Binding Protein Determines Joint-Invading Capacity of Staphylococcus aureus, a Core Mechanism of Septic Arthritis. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Nemecek, D.; Keightley, J.A.; Thomas, G.J., Jr.; Geisbrecht, B.V. The Staphylococcus aureus extracellular adherence protein (Eap) adopts an elongated but structured conformation in solution. Protein Sci. 2007, 16, 2605–2617. [Google Scholar] [CrossRef]
- Zecconi, A.; Scali, F. Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases. Immunol. Lett. 2013, 150, 12–22. [Google Scholar] [CrossRef]
- Kwiecinski, J.; Jacobsson, G.; Karlsson, M.; Zhu, X.; Wang, W.; Bremell, T.; Josefsson, E.; Jin, T. Staphylokinase Promotes the Establishment of Staphylococcus aureus Skin Infections While Decreasing Disease Severity. J. Infect. Dis. 2013, 208, 990–999. [Google Scholar] [CrossRef]
- Xu, S.X.; McCormick, J.K. Staphylococcal superantigens in colonization and disease. Front. Cell. Infect. Microbiol. 2012, 2, 52. [Google Scholar] [CrossRef]
- McCormick, J.K.; Tripp, T.J.; Llera, A.S.; Sundberg, E.J.; Dinges, M.M.; Mariuzza, R.A.; Schlievert, P.M. Functional analysis of the TCR binding domain of toxic shock syndrome toxin-1 predicts further diversity in MHC class II/superantigen/TCR ternary complexes. J. Immunol. 2003, 171, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Abdelnour, A.; Bremell, T.; Holmdahl, R.; Tarkowski, A. Clonal expansion of T lymphocytes causes arthritis and mortality in mice infected with toxic shock syndrome toxin-1-producing staphylococci. Eur. J. Immunol. 1994, 24, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Omoe, K.; Hu, D.L.; Ono, H.K.; Shimizu, S.; Takahashi-Omoe, H.; Nakane, A.; Uchiyama, T.; Shinagawa, K.; Imanishi, K. Emetic potentials of newly identified staphylococcal enterotoxin-like toxins. Infect. Immun. 2013, 81, 3627–3631. [Google Scholar] [CrossRef] [PubMed]
- Vandenesch, F.; Lina, G.; Henry, T. Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: A redundant arsenal of membrane-damaging virulence factors? Front. Cell. Infect. Microbiol. 2012, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.M.; Hartford, O.; Foster, T.; Tarkowski, A. Alpha-toxin and gamma-toxin jointly promote Staphylococcus aureus virulence in murine septic arthritis. Infect. Immun. 1999, 67, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Chan, L.; Tattevin, P.; Kajikawa, O.; Martin, T.R.; Basuino, L.; Mai, T.T.; Marbach, H.; Braughton, K.R.; Whitney, A.R.; et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. USA 2010, 107, 5587–5592. [Google Scholar] [CrossRef]
- Cheung, G.Y.; Joo, H.S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins--critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef]
- Periasamy, S.; Joo, H.S.; Duong, A.C.; Bach, T.H.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286. [Google Scholar] [CrossRef]
- Peschel, A.; Otto, M. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 2013, 11, 667–673. [Google Scholar] [CrossRef]
- Rasigade, J.P.; Trouillet-Assant, S.; Ferry, T.; Diep, B.A.; Sapin, A.; Lhoste, Y.; Ranfaing, J.; Badiou, C.; Benito, Y.; Bes, M.; et al. PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS ONE 2013, 8, e63176. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.M.; Nilsson, I.M.; Verdrengh, M.; Collins, L.V.; Tarkowski, A. Intra-articularly localized bacterial DNA containing CpG motifs induces arthritis. Nat. Med. 1999, 5, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Sparwasser, T.; Miethke, T.; Lipford, G.; Borschert, K.; Hacker, H.; Heeg, K.; Wagner, H. Bacterial DNA causes septic shock. Nature 1997, 386, 336–337. [Google Scholar] [CrossRef]
- Joh, D.; Wann, E.R.; Kreikemeyer, B.; Speziale, P.; Hook, M. Role of fibronectin-binding MSCRAMMs in bacterial adherence and entry into mammalian cells. Matrix Biol. 1999, 18, 211–223. [Google Scholar] [CrossRef]
- An, Y.H.; Friedman, R.J. Handbook of Bacterial Adhesion Principles, Methods, and Applications; Humana Press: Totowa, NJ, USA, 2000; Volume xvi, 645p. [Google Scholar]
- Gomez, M.I.; O’Seaghdha, M.; Magargee, M.; Foster, T.J.; Prince, A.S. Staphylococcus aureus protein A activates TNFR1 signaling through conserved IgG binding domains. J. Biol. Chem. 2006, 281, 20190–20196. [Google Scholar] [CrossRef]
- Chavakis, T.; Wiechmann, K.; Preissner, K.T.; Herrmann, M. Staphylococcus aureus interactions with the endothelium: The role of bacterial “secretable expanded repertoire adhesive molecules” (SERAM) in disturbing host defense systems. Thromb. Haemost. 2005, 94, 278–285. [Google Scholar] [CrossRef]
- Claes, J.; Vanassche, T.; Peetermans, M.; Liesenborghs, L.; Vandenbriele, C.; Vanhoorelbeke, K.; Missiakas, D.; Schneewind, O.; Hoylaerts, M.F.; Heying, R.; et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood 2014, 124, 1669–1676. [Google Scholar] [CrossRef]
- Claes, J.; Liesenborghs, L.; Peetermans, M.; Veloso, T.R.; Missiakas, D.; Schneewind, O.; Mancini, S.; Entenza, J.M.; Hoylaerts, M.F.; Heying, R.; et al. Clumping factor A, von Willebrand factor-binding protein and von Willebrand factor anchor Staphylococcus aureus to the vessel wall. J. Thromb. Haemost. 2017, 15, 1009–1019. [Google Scholar] [CrossRef]
- Bardoel, B.W.; Vos, R.; Bouman, T.; Aerts, P.C.; Bestebroer, J.; Huizinga, E.G.; Brondijk, T.H.; van Strijp, J.A.; de Haas, C.J. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J. Mol. Med. (Berl.) 2012, 90, 1109–1120. [Google Scholar] [CrossRef]
- Ibberson, C.B.; Jones, C.L.; Singh, S.; Wise, M.C.; Hart, M.E.; Zurawski, D.V.; Horswill, A.R. Staphylococcus aureus hyaluronidase is a CodY-regulated virulence factor. Infect. Immun. 2014, 82, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, J.; Peetermans, M.; Liesenborghs, L.; Na, M.; Bjornsdottir, H.; Zhu, X.; Jacobsson, G.; Johansson, B.R.; Geoghegan, J.A.; Foster, T.J.; et al. Staphylokinase Control of Staphylococcus aureus Biofilm Formation and Detachment Through Host Plasminogen Activation. J. Infect. Dis. 2016, 213, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.D. Clarifying the mechanism of superantigen toxicity. PLoS Biol. 2011, 9, e1001145. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Blackman, M.; Kushnir, E.; Kappler, J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J. Exp. Med. 1990, 171, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Kaempfer, R.; Arad, G.; Levy, R.; Hillman, D.; Nasie, I.; Rotfogel, Z. CD28: Direct and critical receptor for superantigen toxins. Toxins (Basel) 2013, 5, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, E. alpha-Hemolysin from Staphylococcus aureus: An archetype of beta-barrel, channel-forming toxins. J. Struct. Biol. 1998, 121, 110–122. [Google Scholar] [CrossRef]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef]
- Deng, G.M.; Liu, Z.Q.; Tarkowski, A. Intracisternally localized bacterial DNA containing CpG motifs induces meningitis. J. Immunol. 2001, 167, 4616–4626. [Google Scholar] [CrossRef]
- Ali, A.; Zhu, X.; Kwiecinski, J.; Gjertsson, I.; Lindholm, C.; Iwakura, Y.; Wang, X.; Lycke, N.; Josefsson, E.; Pullerits, R.; et al. Antibiotic-killed Staphylococcus aureus induces destructive arthritis in mice. Arthritis Rheumatol. 2015, 67, 107–116. [Google Scholar] [CrossRef]
- Verdrengh, M.; Tarkowski, A. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect. Immun. 1997, 65, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Role of macrophages in Staphylococcus aureus-induced arthritis and sepsis. Arthritis Rheum. 2000, 43, 2276–2282. [Google Scholar] [CrossRef]
- Nilsson, N.; Bremell, T.; Tarkowski, A.; Carlsten, H. Protective role of NK1.1+ cells in experimental Staphylococcus aureus arthritis. Clin. Exp. Immunol. 1999, 117, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Krishack, P.A.; Louviere, T.J.; Decker, T.S.; Kuzel, T.G.; Greenberg, J.A.; Camacho, D.F.; Hrusch, C.L.; Sperling, A.I.; Verhoef, P.A. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Abdelnour, A.; Bremell, T.; Holmdahl, R.; Tarkowski, A. Role of T lymphocytes in experimental Staphylococcus aureus arthritis. Scand. J. Immunol. 1994, 39, 403–408. [Google Scholar] [CrossRef]
- Bergmann, B.; Fei, Y.; Jirholt, P.; Hu, Z.; Bergquist, M.; Ali, A.; Lindholm, C.; Ekwall, O.; Churlaud, G.; Klatzmann, D.; et al. Pre-treatment with IL2 gene therapy alleviates Staphylococcus aureus arthritis in mice. BMC Infect. Dis. 2020, 20, 185. [Google Scholar] [CrossRef]
- Gjertsson, I.; Hultgren, O.H.; Stenson, M.; Holmdahl, R.; Tarkowski, A. Are B lymphocytes of importance in severe Staphylococcus aureus infections? Infect. Immun. 2000, 68, 2431–2434. [Google Scholar] [CrossRef]
- Wuescher, L.M.; Takashima, A.; Worth, R.G. A novel conditional platelet depletion mouse model reveals the importance of platelets in protection against Staphylococcus aureus bacteremia. J. Thromb. Haemost. 2015, 13, 303–313. [Google Scholar] [CrossRef]
- Piliponsky, A.M.; Shubin, N.J.; Lahiri, A.K.; Truong, P.; Clauson, M.; Niino, K.; Tsuha, A.L.; Nedospasov, S.A.; Karasuyama, H.; Reber, L.L.; et al. Basophil-derived tumor necrosis factor can enhance survival in a sepsis model in mice. Nat. Immunol. 2019, 20, 129–140. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H. Basic Immunology: Functions and Disorders of the Immune System, 3rd ed.; Saunders/Elsevier: Philadelphia, PA, USA, 2009. [Google Scholar]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Molne, L.; Verdrengh, M.; Tarkowski, A. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect. Immun. 2000, 68, 6162–6167. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. Anatomy of a discovery: m1 and m2 macrophages. Front. Immunol. 2015, 6, 212. [Google Scholar] [CrossRef]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Makita, N.; Hizukuri, Y.; Yamashiro, K.; Murakawa, M.; Hayashi, Y. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. Int. Immunol. 2015, 27, 131–141. [Google Scholar] [CrossRef]
- Dey, A.; Allen, J.; Hankey-Giblin, P.A. Ontogeny and polarization of macrophages in inflammation: Blood monocytes versus tissue macrophages. Front. Immunol. 2014, 5, 683. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakayamada, S.; Okada, Y. Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 325–328. [Google Scholar] [CrossRef]
- Ogata, Y.; Kukita, A.; Kukita, T.; Komine, M.; Miyahara, A.; Miyazaki, S.; Kohashi, O. A novel role of IL-15 in the development of osteoclasts: Inability to replace its activity with IL-2. J. Immunol. 1999, 162, 2754–2760. [Google Scholar]
- Djaafar, S.; Pierroz, D.D.; Chicheportiche, R.; Zheng, X.X.; Ferrari, S.L.; Ferrari-Lacraz, S. Inhibition of T cell-dependent and RANKL-dependent osteoclastogenic processes associated with high levels of bone mass in interleukin-15 receptor-deficient mice. Arthritis Rheum. 2010, 62, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Henningsson, L.; Jirholt, P.; Bogestal, Y.R.; Eneljung, T.; Adiels, M.; Lindholm, C.; McInnes, I.; Bulfone-Paus, S.; Lerner, U.H.; Gjertsson, I. Interleukin 15 mediates joint destruction in Staphylococcus aureus arthritis. J. Infect. Dis. 2012, 206, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Bokarewa, M.; Ohlsson, C.; Stolina, M.; Tarkowski, A. RANKL-targeted therapy inhibits bone resorption in experimental Staphylococcus aureus-induced arthritis. Bone 2010, 46, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.M.; Verdrengh, M.; Liu, Z.Q.; Tarkowski, A. The major role of macrophages and their product tumor necrosis factor alpha in the induction of arthritis triggered by bacterial DNA containing CpG motifs. Arthritis Rheum. 2000, 43, 2283–2289. [Google Scholar] [CrossRef]
- Small, C.L.; McCormick, S.; Gill, N.; Kugathasan, K.; Santosuosso, M.; Donaldson, N.; Heinrichs, D.E.; Ashkar, A.; Xing, Z. NK cells play a critical protective role in host defense against acute extracellular Staphylococcus aureus bacterial infection in the lung. J. Immunol. 2008, 180, 5558–5568. [Google Scholar] [CrossRef]
- Laarman, A.J.; Ruyken, M.; Malone, C.L.; van Strijp, J.A.; Horswill, A.R.; Rooijakkers, S.H. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J. Immunol. 2011, 186, 6445–6453. [Google Scholar] [CrossRef]
- Laarman, A.; Milder, F.; van Strijp, J.; Rooijakkers, S. Complement inhibition by gram-positive pathogens: Molecular mechanisms and therapeutic implications. J. Mol. Med. (Berl.) 2010, 88, 115–120. [Google Scholar] [CrossRef]
- Sakiniene, E.; Bremell, T.; Tarkowski, A. Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin. Exp. Immunol. 1999, 115, 95–102. [Google Scholar] [CrossRef]
- Na, M.; Jarneborn, A.; Ali, A.; Welin, A.; Magnusson, M.; Stokowska, A.; Pekna, M.; Jin, T. Deficiency of the complement component 3 but not factor B aggravates Staphylococcus aureus septic arthritis in mice. Infect. Immun. 2016. [Google Scholar] [CrossRef]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Kidd, P. Th1/Th2 balance: The hypothesis, its limitations, and implications for health and disease. Altern. Med. Rev. 2003, 8, 223–246. [Google Scholar] [PubMed]
- Romagnani, S. Th1/Th2 cells. Inflamm. Bowel Dis. 1999, 5, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Oosterwegel, M.A.; Mandelbrot, D.A.; Boyd, S.D.; Lorsbach, R.B.; Jarrett, D.Y.; Abbas, A.K.; Sharpe, A.H. The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 1999, 163, 2634–2639. [Google Scholar] [PubMed]
- Ali, A.; Welin, A.; Schwarze, J.C.; Svensson, M.N.; Na, M.; Jarneborn, A.; Magnusson, M.; Mohammad, M.; Kwiecinski, J.; Josefsson, E.; et al. CTLA4 Immunoglobulin but Not Anti-Tumor Necrosis Factor Therapy Promotes Staphylococcal Septic Arthritis in Mice. J. Infect. Dis. 2015, 212, 1308–1316. [Google Scholar] [CrossRef]
- Parker, D.; Ryan, C.L.; Alonzo, F., 3rd; Torres, V.J.; Planet, P.J.; Prince, A.S. CD4+ T cells promote the pathogenesis of Staphylococcus aureus pneumonia. J. Infect. Dis. 2015, 211, 835–845. [Google Scholar] [CrossRef]
- Jin, T.; Mohammad, M.; Hu, Z.; Fei, Y.; Moore, E.R.B.; Pullerits, R.; Ali, A. A novel mouse model for septic arthritis induced by Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 16868. [Google Scholar] [CrossRef]
- Cho, J.S.; Pietras, E.M.; Garcia, N.C.; Ramos, R.I.; Farzam, D.M.; Monroe, H.R.; Magorien, J.E.; Blauvelt, A.; Kolls, J.K.; Cheung, A.L.; et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Investig. 2010, 120, 1762–1773. [Google Scholar] [CrossRef]
- Henningsson, L.; Jirholt, P.; Lindholm, C.; Eneljung, T.; Silverpil, E.; Iwakura, Y.; Linden, A.; Gjertsson, I. Interleukin-17A during local and systemic Staphylococcus aureus-induced arthritis in mice. Infect. Immun. 2010, 78, 3783–3790. [Google Scholar] [CrossRef]
- Kwiecinski, J.; Rhost, S.; Lofbom, L.; Blomqvist, M.; Mansson, J.E.; Cardell, S.L.; Jin, T. Sulfatide attenuates experimental Staphylococcus aureus sepsis through a CD1d-dependent pathway. Infect. Immun. 2013, 81, 1114–1120. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Hepworth, M.R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 2019, 19, 599–613. [Google Scholar] [CrossRef]
- Ali, R.A.; Wuescher, L.M.; Dona, K.R.; Worth, R.G. Platelets Mediate Host Defense against Staphylococcus aureus through Direct Bactericidal Activity and by Enhancing Macrophage Activities. J. Immunol. 2017, 198, 344–351. [Google Scholar] [CrossRef]
- Hultgren, O.; Eugster, H.P.; Sedgwick, J.D.; Korner, H.; Tarkowski, A. TNF/lymphotoxin-alpha double-mutant mice resist septic arthritis but display increased mortality in response to Staphylococcus aureus. J. Immunol. 1998, 161, 5937–5942. [Google Scholar]
- Hultgren, O.H.; Svensson, L.; Tarkowski, A. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J. Immunol. 2002, 168, 5207–5212. [Google Scholar] [CrossRef]
- Lenski, M.; Scherer, M.A. The significance of interleukin-6 and lactate in the synovial fluid for diagnosing native septic arthritis. Acta Orthop. Belg. 2014, 80, 18–25. [Google Scholar]
- Hultgren, O.H.; Stenson, M.; Tarkowski, A. Role of IL-12 in Staphylococcus aureus-triggered arthritis and sepsis. Arthritis Res. 2001, 3, 41–47. [Google Scholar] [CrossRef]
- Hultgren, O.; Kopf, M.; Tarkowski, A. Staphylococcus aureus-induced septic arthritis and septic death is decreased in IL-4-deficient mice: Role of IL-4 as promoter for bacterial growth. J. Immunol. 1998, 160, 5082–5087. [Google Scholar]
- Hultgren, O.; Kopf, M.; Tarkowski, A. Outcome of Staphylococcus aureus-triggered sepsis and arthritis in IL-4-deficient mice depends on the genetic background of the host. Eur. J. Immunol. 1999, 29, 2400–2405. [Google Scholar] [CrossRef]
- Gjertsson, I.; Hultgren, O.H.; Tarkowski, A. Interleukin-10 ameliorates the outcome of Staphylococcus aureus arthritis by promoting bacterial clearance. Clin. Exp. Immunol. 2002, 130, 409–414. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Nilsson, I.M.; Tarkowski, A. The dual role of interferon-gamma in experimental Staphylococcus aureus septicaemia versus arthritis. Immunology 1998, 93, 80–85. [Google Scholar] [CrossRef]
- Jarneborn, A.; Mohammad, M.; Engdahl, C.; Hu, Z.; Na, M.; Ali, A.; Jin, T. Tofacitinib treatment aggravates Staphylococcus aureus septic arthritis, but attenuates sepsis and enterotoxin induced shock in mice. Sci. Rep. 2020, 10, 10891. [Google Scholar] [CrossRef]
- Davis, L.S.; Hutcheson, J.; Mohan, C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J. Interferon Cytokine Res. 2011, 31, 781–789. [Google Scholar] [CrossRef]
- Osiri, M.; Ruxrungtham, K.; Nookhai, S.; Ohmoto, Y.; Deesomchok, U. IL-1beta, IL-6 and TNF-alpha in synovial fluid of patients with non-gonococcal septic arthritis. Asian Pac. J. Allergy Immunol. 1998, 16, 155–160. [Google Scholar]
- Na, M.; Wang, W.; Fei, Y.; Josefsson, E.; Ali, A.; Jin, T. Both anti-TNF and CTLA4 Ig treatments attenuate the disease severity of staphylococcal dermatitis in mice. PLoS ONE 2017, 12, e0173492. [Google Scholar] [CrossRef]
- Jacques, C.; Gosset, M.; Berenbaum, F.; Gabay, C. The role of IL-1 and IL-1Ra in joint inflammation and cartilage degradation. Vitam. Horm. 2006, 74, 371–403. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Tarkowski, A. Impact of interferon-gamma receptor deficiency on experimental Staphylococcus aureus septicemia and arthritis. J. Immunol. 1995, 155, 5736–5742. [Google Scholar]
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- Ivanov, S.; Linden, A. Interleukin-17 as a drug target in human disease. Trends Pharmacol. Sci. 2009, 30, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Sharff, K.A.; Richards, E.P.; Townes, J.M. Clinical management of septic arthritis. Curr. Rheumatol. Rep. 2013, 15, 332. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, A. Infection and musculoskeletal conditions: Infectious arthritis. Best Pract. Res. Clin. Rheumatol. 2006, 20, 1029–1044. [Google Scholar] [CrossRef]
- Goldenberg, D.L. Septic arthritis. Lancet 1998, 351, 197–202. [Google Scholar] [CrossRef]
- Garcia-Arias, M.; Balsa, A.; Mola, E.M. Septic arthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 407–421. [Google Scholar] [CrossRef]
- Mathews, C.J.; Weston, V.C.; Jones, A.; Field, M.; Coakley, G. Bacterial septic arthritis in adults. Lancet 2010, 375, 846–855. [Google Scholar] [CrossRef]
- Morgan, D.S.; Fisher, D.; Merianos, A.; Currie, B.J. An 18 year clinical review of septic arthritis from tropical Australia. Epidemiol. Infect. 1996, 117, 423–428. [Google Scholar] [CrossRef]
- Fowler, V.G., Jr.; Sanders, L.L.; Sexton, D.J.; Kong, L.; Marr, K.A.; Gopal, A.K.; Gottlieb, G.; McClelland, R.S.; Corey, G.R. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendations of infectious diseases specialists: Experience with 244 patients. Clin. Infect. Dis. 1998, 27, 478–486. [Google Scholar] [CrossRef]
- Marr, K.A.; Kong, L.; Fowler, V.G.; Gopal, A.; Sexton, D.J.; Conlon, P.J.; Corey, G.R. Incidence and outcome of Staphylococcus aureus bacteremia in hemodialysis patients. Kidney Int. 1998, 54, 1684–1689. [Google Scholar] [CrossRef][Green Version]
- Naber, C.K. Staphylococcus aureus bacteremia: Epidemiology, pathophysiology, and management strategies. Clin. Infect. Dis. 2009, 48 (Suppl. 4), S231–S237. [Google Scholar] [CrossRef]
- Holland, T.L.; Arnold, C.; Fowler, V.G., Jr. Clinical management of Staphylococcus aureus bacteremia: A review. JAMA 2014, 312, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S. Sepsis, severe sepsis and septic shock: Changes in incidence, pathogens and outcomes. Expert. Rev. Anti Infect. Ther. 2012, 10, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Van Amersfoort, E.S.; Van Berkel, T.J.; Kuiper, J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 2003, 16, 379–414. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, R.; Aittoniemi, J. New concepts in the pathogenesis, diagnosis and treatment of bacteremia and sepsis. J. Infect. 2011, 63, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, N.; Ammollo, C.T.; Semeraro, F.; Colucci, M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr. J. Hematol. Infect. Dis. 2010, 2, e2010024. [Google Scholar] [CrossRef] [PubMed]
- Romling, U.; Balsalobre, C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J. Intern. Med. 2012, 272, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, P.; Nistico, L.; Johnson, S.; Lasko, L.A.; Baratz, M.; Gahlot, V.; Ehrlich, G.D.; Kathju, S. Direct demonstration of viable Staphylococcus aureus biofilms in an infected total joInt. arthroplasty. A case report. J. Bone Joint Surg. Am. 2008, 90, 1751–1758. [Google Scholar] [CrossRef]
- Dastgheyb, S.; Parvizi, J.; Shapiro, I.M.; Hickok, N.J.; Otto, M. Effect of biofilms on recalcitrance of staphylococcal joInt. infection to antibiotic treatment. J. Infect. Dis. 2015, 211, 641–650. [Google Scholar] [CrossRef]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Patti, J.M.; Allen, B.L.; McGavin, M.J.; Hook, M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 1994, 48, 585–617. [Google Scholar] [CrossRef]
- Kwiecinski, J.; Kahlmeter, G.; Jin, T. Biofilm formation by Staphylococcus aureus isolates from skin and soft tissue infections. Curr. Microbiol. 2015, 70, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcal infections: Mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu. Rev. Med. 2013, 64, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, J.; Na, M.; Jarneborn, A.; Jacobsson, G.; Peetermans, M.; Verhamme, P.; Jin, T. Tissue Plasminogen Activator Coating on Implant Surfaces Reduces Staphylococcus aureus Biofilm Formation. Appl. Environ. Microbiol. 2015, 82, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, Z.J.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef]
- Libertucci, J.; Young, V.B. The role of the microbiota in infectious diseases. Nat. Microbiol. 2019, 4, 35–45. [Google Scholar] [CrossRef]
- Sewankambo, N.; Gray, R.H.; Wawer, M.J.; Paxton, L.; McNaim, D.; Wabwire-Mangen, F.; Serwadda, D.; Li, C.; Kiwanuka, N.; Hillier, S.L.; et al. HIV-1 infection associated with abnormal vaginal flora morphology and bacterial vaginosis. Lancet 1997, 350, 546–550. [Google Scholar] [CrossRef]
- Hyoju, S.K.; Zaborin, A.; Keskey, R.; Sharma, A.; Arnold, W.; van den Berg, F.; Kim, S.M.; Gottel, N.; Bethel, C.; Charnot-Katsikas, A.; et al. Mice Fed an Obesogenic Western Diet, Administered Antibiotics, and Subjected to a Sterile Surgical Procedure Develop Lethal Septicemia with Multidrug-Resistant Pathobionts. mBio 2019, 10. [Google Scholar] [CrossRef]
- Prescott, H.C.; Dickson, R.P.; Rogers, M.A.; Langa, K.M.; Iwashyna, T.J. Hospitalization Type and Subsequent Severe Sepsis. Am. J. Respir. Crit. Care Med. 2015, 192, 581–588. [Google Scholar] [CrossRef]
- Taur, Y.; Xavier, J.B.; Lipuma, L.; Ubeda, C.; Goldberg, J.; Gobourne, A.; Lee, Y.J.; Dubin, K.A.; Socci, N.D.; Viale, A.; et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 2012, 55, 905–914. [Google Scholar] [CrossRef]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Jubair, W.K.; Hendrickson, J.D.; Severs, E.L.; Schulz, H.M.; Adhikari, S.; Ir, D.; Pagan, J.D.; Anthony, R.M.; Robertson, C.E.; Frank, D.N.; et al. Modulation of Inflammatory Arthritis in Mice by Gut Microbiota Through Mucosal Inflammation and Autoantibody Generation. Arthritis Rheumatol. 2018, 70, 1220–1233. [Google Scholar] [CrossRef]
- Breban, M.; Tap, J.; Leboime, A.; Said-Nahal, R.; Langella, P.; Chiocchia, G.; Furet, J.P.; Sokol, H. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Ulici, V.; Kelley, K.L.; Azcarate-Peril, M.A.; Cleveland, R.J.; Sartor, R.B.; Schwartz, T.A.; Loeser, R.F. Osteoarthritis induced by destabilization of the medial meniscus is reduced in germ-free mice. Osteoarthr. Cartil. 2018, 26, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.; Patrier, S.; Pralus, A.; Roche, M.; Nivoliez, A. Protective Actions of Oral Administration of Bifidobacterium longum CBi0703 in Spontaneous Osteoarthritis in Dunkin Hartley Guinea Pig Model. Cartilage 2019, 1947603519841674. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.L.; Bomhof, M.R.; Reimer, R.A.; Hart, D.A.; Collins, K.H.; Herzog, W. Protective effect of prebiotic and exercise intervention on knee health in a rat model of diet-induced obesity. Sci. Rep. 2019, 9, 3893. [Google Scholar] [CrossRef] [PubMed]
- Kaandorp, C.J.; Van Schaardenburg, D.; Krijnen, P.; Habbema, J.D.; van de Laar, M.A. Risk factors for septic arthritis in patients with joint disease. A prospective study. Arthritis Rheum. 1995, 38, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.C.; Gravallese, E.M. Bone remodeling in rheumatic disease: A question of balance. Immunol. Rev. 2010, 233, 301–312. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef]
- Fei, Y.; Wang, W.; Kwiecinski, J.; Josefsson, E.; Pullerits, R.; Jonsson, I.M.; Magnusson, M.; Jin, T. The combination of a tumor necrosis factor inhibitor and antibiotic alleviates staphylococcal arthritis and sepsis in mice. J. Infect. Dis. 2011, 204, 348–357. [Google Scholar] [CrossRef]
- Ali, A.; Na, M.; Svensson, M.N.; Magnusson, M.; Welin, A.; Schwarze, J.C.; Mohammad, M.; Josefsson, E.; Pullerits, R.; Jin, T. IL-1 Receptor Antagonist Treatment Aggravates Staphylococcal Septic Arthritis and Sepsis in Mice. PLoS ONE 2015, 10, e0131645. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Fong, Y.; Hesse, D.G.; Manogue, K.R.; Lee, A.T.; Kuo, G.C.; Lowry, S.F.; Cerami, A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987, 330, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Okusawa, S.; Gelfand, J.A.; Ikejima, T.; Connolly, R.J.; Dinarello, C.A. Interleukin 1 induces a shock-like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J. Clin. Investig. 1988, 81, 1162–1172. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.J., Jr.; Agosti, J.M.; Opal, S.M.; Lowry, S.F.; Balk, R.A.; Sadoff, J.C.; Abraham, E.; Schein, R.M.; Benjamin, E. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N. Engl. J. Med. 1996, 334, 1697–1702. [Google Scholar] [CrossRef]
- Richter, A.; Listing, J.; Schneider, M.; Klopsch, T.; Kapelle, A.; Kaufmann, J.; Zink, A.; Strangfeld, A. Impact of treatment with biologic DMARDs on the risk of sepsis or mortality after serious infection in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, F.; Ward, P.A. Understanding Immunosuppression after Sepsis. Immunity 2017, 47, 3–5. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef]
- Miller, L.S.; Fowler, V.G.; Shukla, S.K.; Rose, W.E.; Proctor, R.A. Development of a vaccine against Staphylococcus aureus invasive infections: Evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol. Rev. 2020, 44, 123–153. [Google Scholar] [CrossRef]
- Smith, R.L.; Kajiyama, G.; Schurman, D.J. Staphylococcal septic arthritis: Antibiotic and nonsteroidal anti-inflammatory drug treatment in a rabbit model. J. Orthop. Res. 1997, 15, 919–926. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Xia, Z.J.; Clement, A.; Parry, D.A.; Davids, M.; Taylan, F.; Sharma, P.; Turgeon, C.T.; Blanco-Sanchez, B.; Ng, B.G.; et al. A Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation. Am. J. Hum. Genet. 2018, 103, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Sakiniene, E.; Bremell, T.; Tarkowski, A. Addition of corticosteroids to antibiotic treatment ameliorates the course of experimental Staphylococcus aureus arthritis. Arthritis Rheum. 1996, 39, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Odio, C.M.; Ramirez, T.; Arias, G.; Abdelnour, A.; Hidalgo, I.; Herrera, M.L.; Bolanos, W.; Alpizar, J.; Alvarez, P. Double blind, randomized, placebo-controlled study of dexamethasone therapy for hematogenous septic arthritis in children. Pediatr. Infect. Dis. J. 2003, 22, 883–888. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Biological Functions | Role in Infection | |
|---|---|---|
| Cell Wall Components: | ||
| Capsular Polysaccharide | Antiphagocytic [4] | Septic arthritis, sepsis [5] |
| Peptidoglycan | Release of TNF-α, IL-6 [12] | Arthritis [12], bone loss [13] |
| Lipoproteins | Activation of innate immunity [25] | Arthritis [14], sepsis [27] |
| Surface Proteins: | ||
| ClfA | Inhibit complement-mediated phagocytosis [29,30] | Septic arthritis, sepsis [31] |
| SpA | Inhibit opsonophagocytosis, B-cell superantigen [32] | Septic arthritis, sepsis [33] |
| FnBPs | Adhesion and invasion of cells [29] | Biofilm formation [29], sepsis [34] |
| Cna | Mediates binding of S. aureus to cartilage [35] | Septic arthritis [36] |
| Secreted Proteins: | ||
| vWbp | Promote blood-clotting [37] | Septic arthritis [38] |
| Eap | Adhesin [39,40] | Inhibit wound healing, biofilm formation [39,40] |
| Staphylokinase | Mediates digestion of fibrin clots [41] | S. aureus skin infections [41] |
| Toxins: | ||
| TSST-1 | Superantigen [42] | Toxic shock syndrome [43], septic arthritis [44] |
| SE | Superantigen [42] | Toxic shock syndrome [45], food poisoning [46] |
| SEls | Superantigen [42] | Food poisoning [46] |
| α- and γ-toxins | Cell lysis [47] | Septic arthritis [48] |
| PVL | Cell lysis [47] | Necrotizing pneumonia [49] |
| PSMs | Cell lysis [47], induce inflammation [50] | Biofilm formation [50,51,52], osteomyelitis [53] |
| Bacterial DNA | Release of TNF-α, IL-6, IFN-γ [54] | Arthritis [55], septic shock [56] |
| Cell Types | Septic Arthritis | Bacterial Clearance | Sepsis |
|---|---|---|---|
| Neutrophils | Protective [74] | Enhance [74] | Pathogenic [74] |
| Monocytes/macrophages | Pathogenic [75] | Enhance [75] | Protective [75] |
| NK cells | Protective [76] | Enhance [76] | Protective [76] |
| Innate lymphoid cells | NA | NA | Type 2 ILCs are protective [77] |
| CD4+ T cells | Pathogenic [78] | No effect [78] | Pathogenic [78] |
| CD8 T cells | NA | NA | NA |
| Regulatory T cells | Protective [79] | No effect [79] | NA |
| Th17 T cells | NA | NA | NA |
| B cells | No effect [80] | No effect [80] | Protective? [80] |
| Thrombocytes | NA | NA | Protective [81] |
| Eosinophils | NA | NA | Protective [77] |
| Basophils | NA | NA | Protective [82] |
| Cytokine | Cell Source | Function | Role in S. aureus Infections |
|---|---|---|---|
| TNF-α | Macrophages T-cells | Proinflammatory | Aggravate S. aureus induced septic arthritis but protective in sepsis [118]. |
| IL-1 | Macrophages Dendritic cells Endothelial cells | Proinflammatory | Protective in S. aureus induced septic arthritis and sepsis [119]. |
| IL-6 | Macrophages and T cells | Proinflammatory | Elevated IL-6 levels in synovial fluid from septic arthritis patients [120]. The role of IL-6 was not yet assessed in animal models for septic arthritis and sepsis. |
| IL-12 | Monocytes Macrophages Dendritic cells | Proinflammatory | Protective in S. aureus induced sepsis but not septic arthritis [121] |
| IL-4 | Th2 cells | Anti-inflammatory | Dual role in S. aureus induced septic arthritis and sepsis depending on the genetic background of the host [122,123]. |
| IL-10 | Monocytes Dendritic cells T-cells | Anti-inflammatory | Protective in S. aureus induced septic arthritis [124]. |
| IL-17 | Th17 cells | Proinflammatory | Protective in local but not systemic S. aureus infection [114]. |
| IFN-γ | NK cells T-cells | Proinflammatory | Protective in S. aureus induced sepsis but aggravates septic arthritis [125]. |
| Janus kinase | All cells | Proinflammatory | Protective in S. aureus septic arthritis but pathogenic in sepsis [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, T.; Mohammad, M.; Pullerits, R.; Ali, A. Bacteria and Host Interplay in Staphylococcus aureus Septic Arthritis and Sepsis. Pathogens 2021, 10, 158. https://doi.org/10.3390/pathogens10020158
Jin T, Mohammad M, Pullerits R, Ali A. Bacteria and Host Interplay in Staphylococcus aureus Septic Arthritis and Sepsis. Pathogens. 2021; 10(2):158. https://doi.org/10.3390/pathogens10020158
Chicago/Turabian StyleJin, Tao, Majd Mohammad, Rille Pullerits, and Abukar Ali. 2021. "Bacteria and Host Interplay in Staphylococcus aureus Septic Arthritis and Sepsis" Pathogens 10, no. 2: 158. https://doi.org/10.3390/pathogens10020158
APA StyleJin, T., Mohammad, M., Pullerits, R., & Ali, A. (2021). Bacteria and Host Interplay in Staphylococcus aureus Septic Arthritis and Sepsis. Pathogens, 10(2), 158. https://doi.org/10.3390/pathogens10020158