
Distribution of ermB, ermF, tet(W), and tet(M) Resistance Genes in the Vaginal Ecosystem of Women during Pregnancy and Puerperium

 , ,
, ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Study Population and Samples
2.2. Detection of Resistance Genes
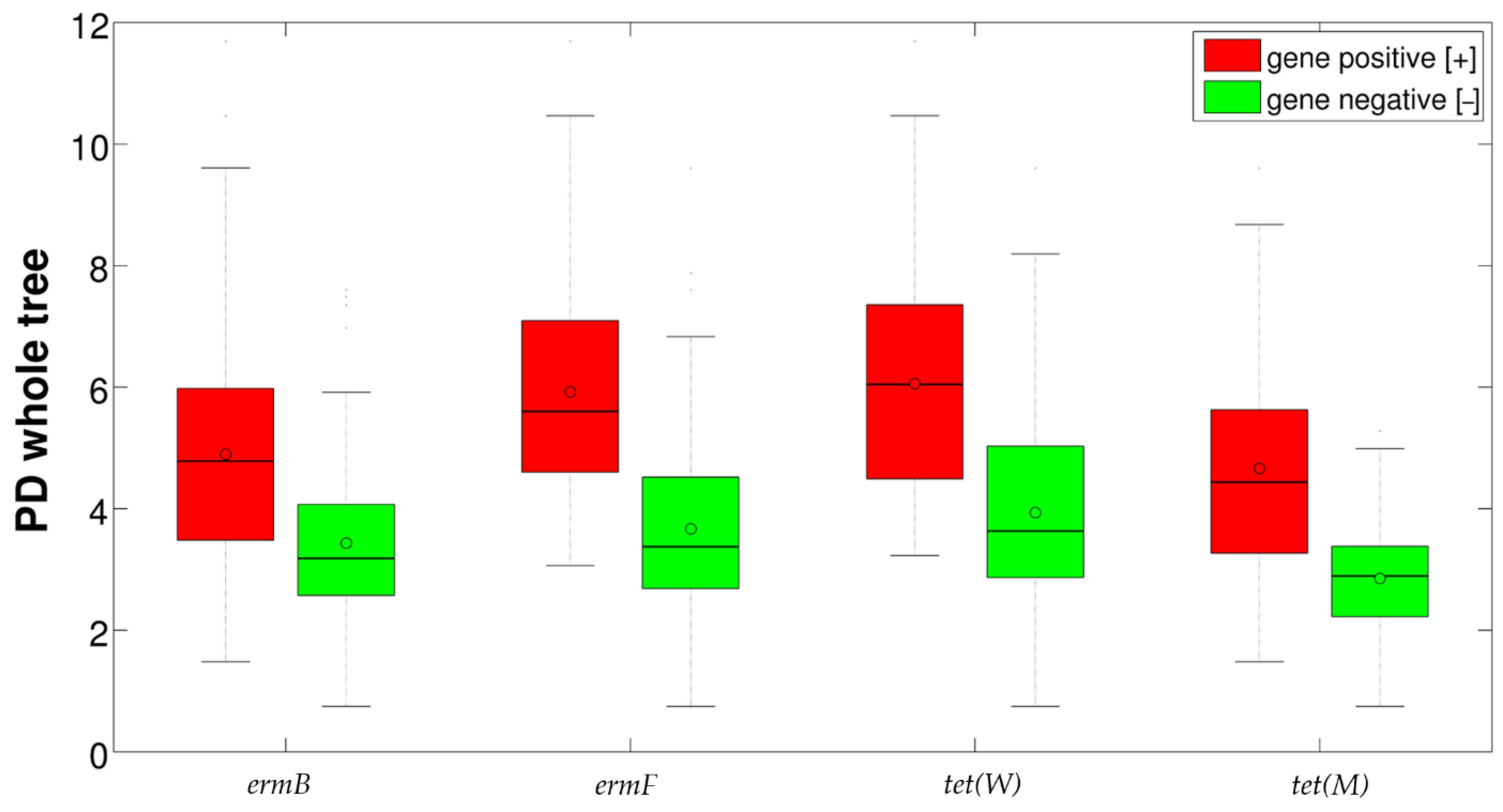
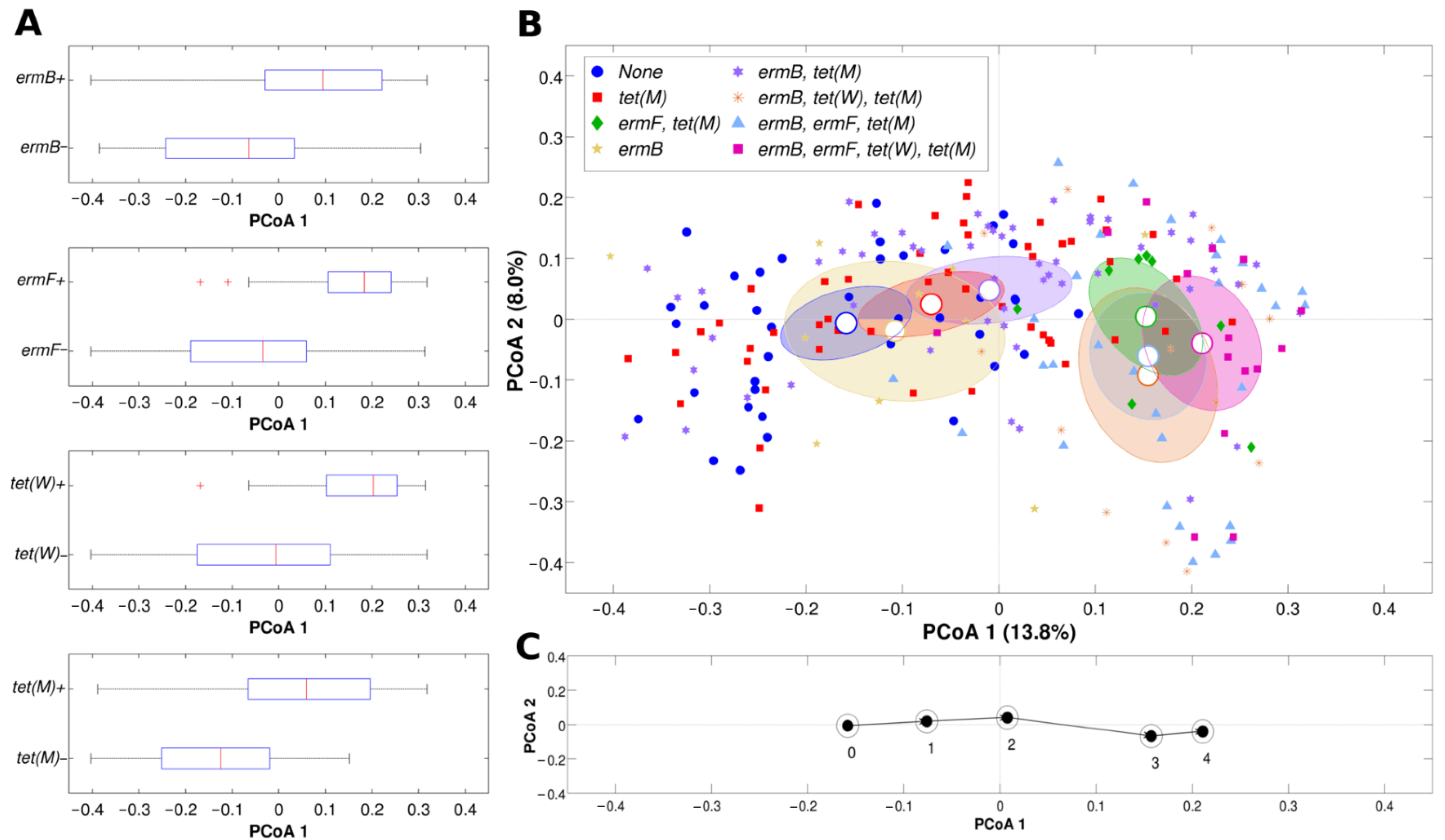
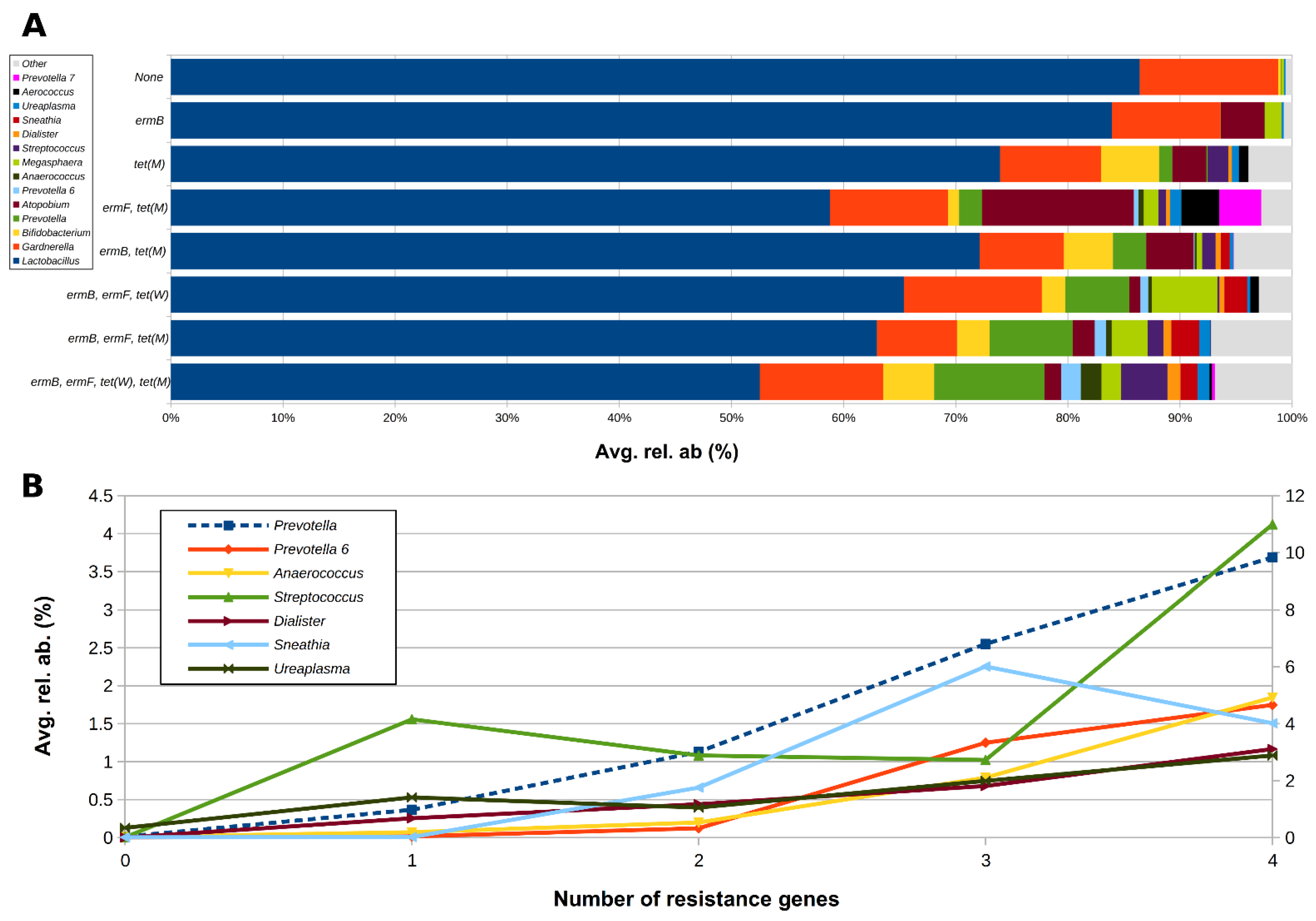
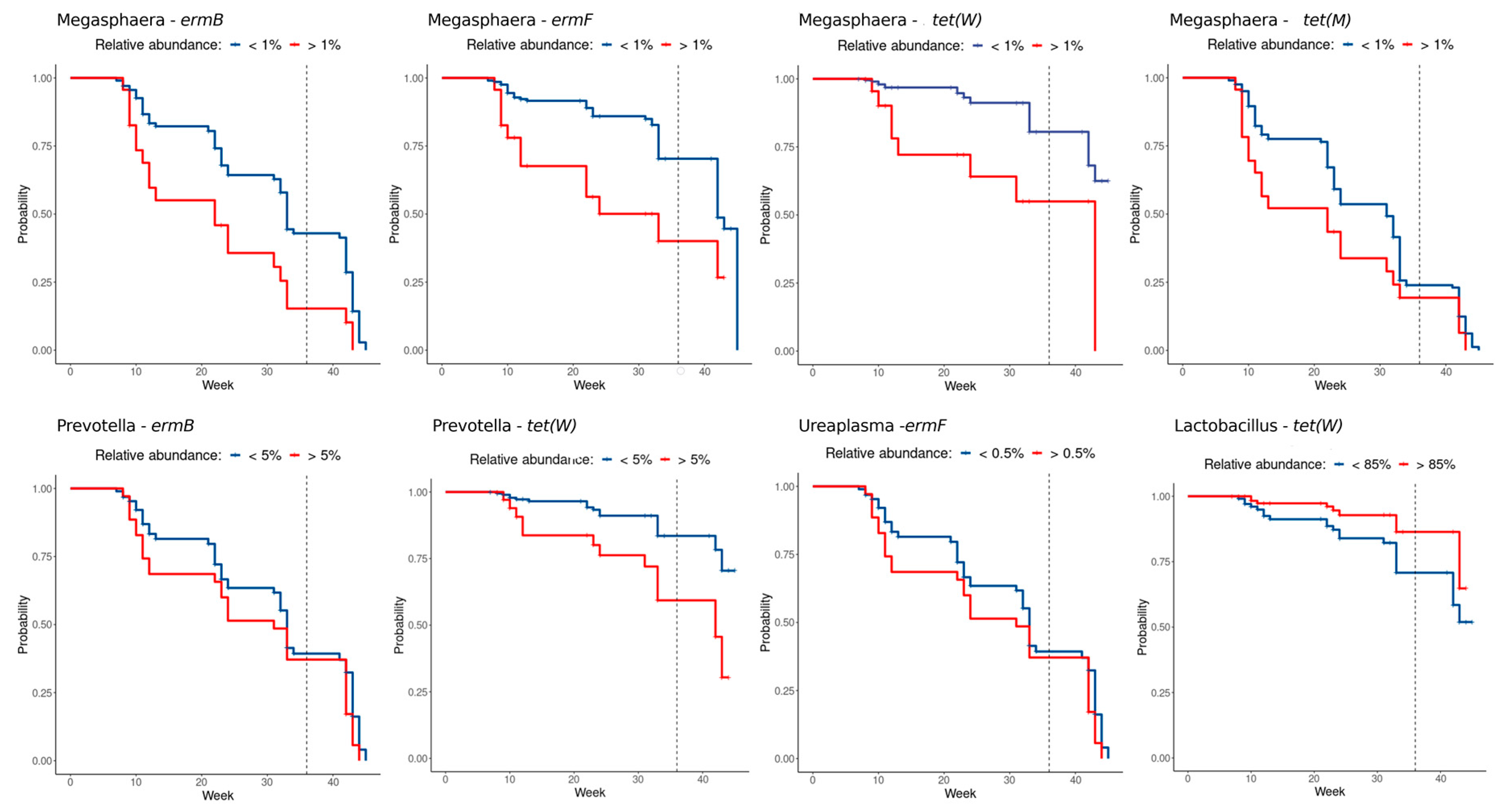
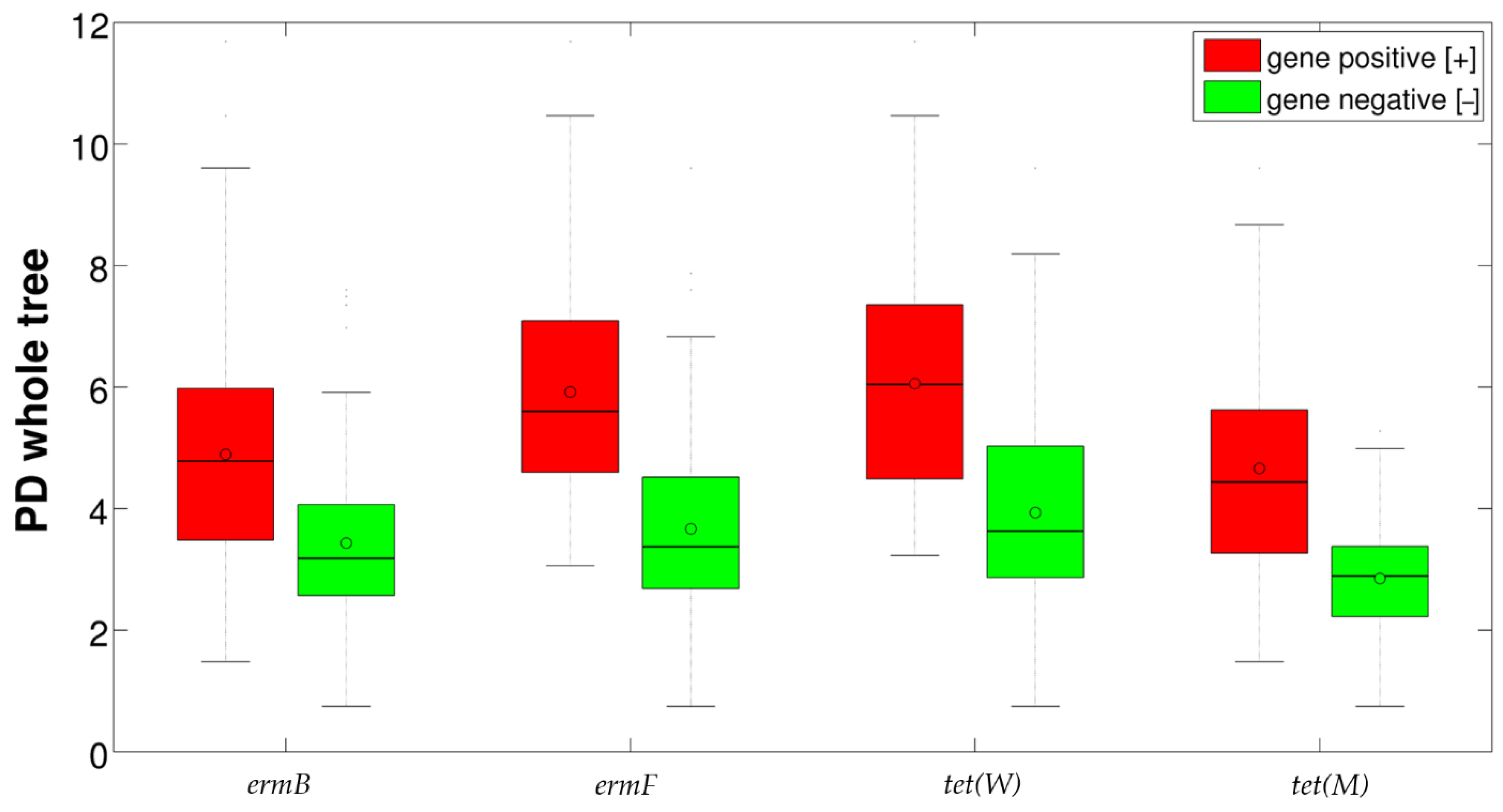
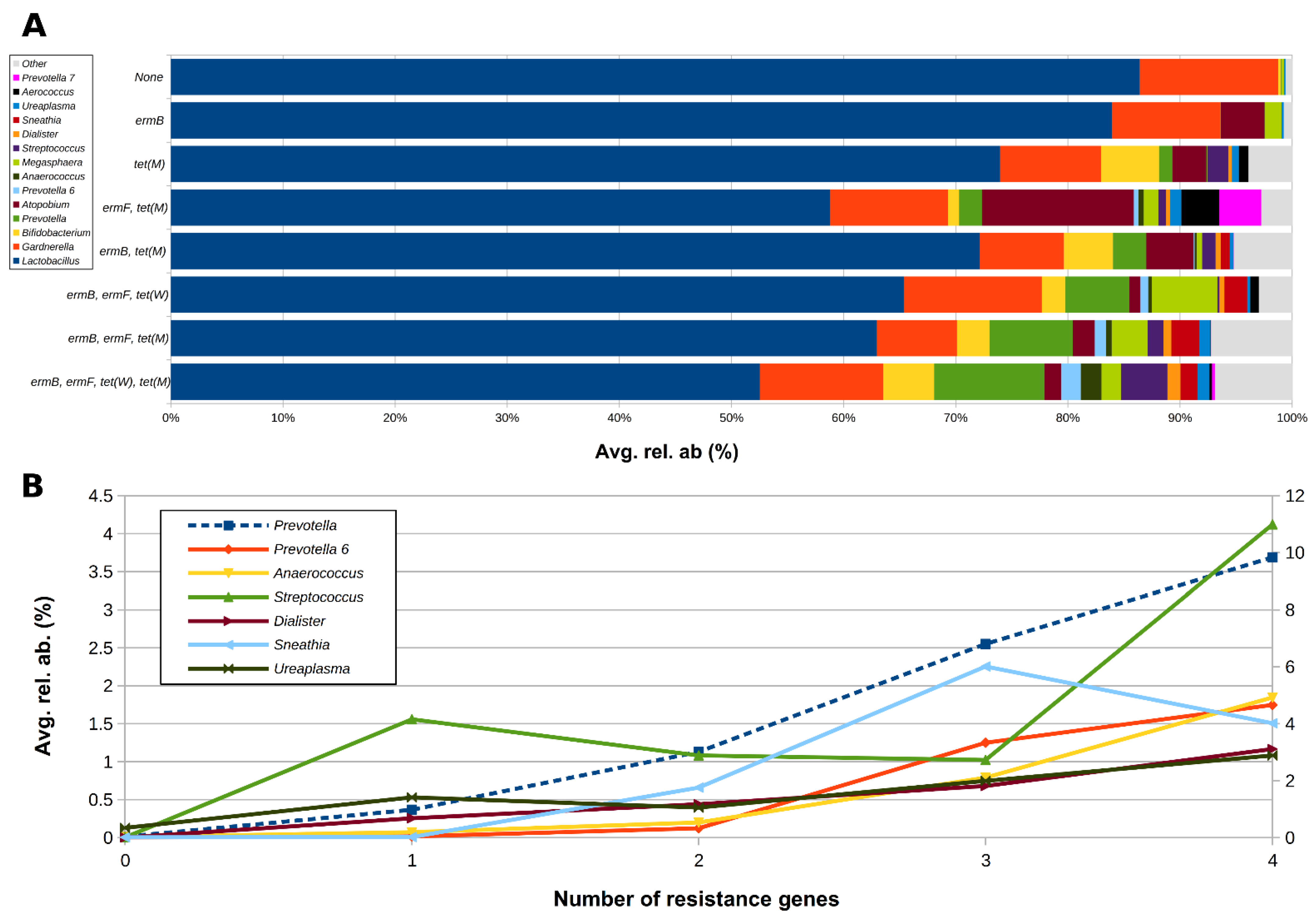
2.3. Correlation between Resistance Genes and Vaginal Microbiota
2.4. Correlation between BMI and Vaginal Microbiota Composition
3. Discussion
4. Materials and Methods
4.1. Study Population and Sample Collection
4.2. Microbiological Investigations
4.3. Detection of Resistance Genes
4.4. Microbiota Analysis
4.5. Statistical Method
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, S.B.; Ravel, J. The vaginal microbiota, host defence and reproductive physiology. J. Physiol. 2017, 595, 451–463. [Google Scholar] [CrossRef] [Green Version]
- van de Wijgert, J.H.; Borgdorff, H.; Verhelst, R.; Crucitti, T.; Francis, S.; Verstraelen, H.; Jespers, V. The vaginal microbiota: What have we learned after a decade of molecular characterization? PLoS ONE 2014, 9, e105998. [Google Scholar] [CrossRef] [Green Version]
- Parolin, C.; Marangoni, A.; Laghi, L.; Foschi, C.; Ñahui Palomino, R.A.; Calonghi, N.; Cevenini, R.; Vitali, B. Isolation of Vaginal Lactobacilli and Characterization of Anti-Candida Activity. PLoS ONE 2015, 10, e0131220. [Google Scholar] [CrossRef] [PubMed]
- Foschi, C.; Salvo, M.; Cevenini, R.; Parolin, C.; Vitali, B.; Marangoni, A. Vaginal lactobacilli reduce Neisseria gonorrhoeae viability through multiple strategies: An in vitro study. Front. Cell Infect. Microbiol. 2017, 7, 502. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Kong, J.; Liu, F.; Zhu, H.; Chen, X.; Wang, Y.; Li, L.; Nelson, K.E.; Xia, Y.; Xiang, C. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genom. 2010, 11, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auriemma, R.S.; Scairati, R.; Del Vecchio, G.; Liccardi, A.; Verde, N.; Pirchio, R.; Pivonello, R.; Ercolini, D.; Colao, A. The Vaginal Microbiome: A Long Urogenital Colonization Throughout Woman Life. Front. Cell Infect. Microbiol. 2021, 11, 686167. [Google Scholar] [CrossRef] [PubMed]
- Parolin, C.; Foschi, C.; Laghi, L.; Zhu, C.; Banzola, N.; Gaspari, V.; D’Antuono, A.; Giordani, B.; Severgnini, M.; Consolandi, C.; et al. Insights Into Vaginal Bacterial Communities and Metabolic Profiles of Chlamydia trachomatis Infection: Positioning Between Eubiosis and Dysbiosis. Front. Microbiol. 2018, 9, 600. [Google Scholar] [CrossRef]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [Green Version]
- Noyes, N.; Cho, K.C.; Ravel, J.; Forney, L.J.; Abdo, Z. Associations between sexual habits, menstrual hygiene practices, demographics and the vaginal microbiome as revealed by Bayesian network analysis. PLoS ONE 2018, 13, e0191625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’Asta, M.; Laghi, L.; Morselli, S.; Re, M.C.; Zagonari, S.; Patuelli, G.; Foschi, C.; Pedna, M.F.; Sambri, V.; Marangoni, A.; et al. Pre-Pregnancy Diet and Vaginal Environment in Caucasian Pregnant Women: An Exploratory Study. Front. Mol. Biosci. 2021, 8, 702370. [Google Scholar] [CrossRef]
- Gupta, P.; Singh, M.P.; Goyal, K. Diversity of Vaginal Microbiome in Pregnancy: Deciphering the Obscurity. Front. Public Health 2020, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, A.; Laghi, L.; Zagonari, S.; Patuelli, G.; Zhu, C.; Foschi, C.; Morselli, S.; Pedna, M.F.; Sambri, V. New Insights into Vaginal Environment During Pregnancy. Front. Mol. Biosci. 2021, 8, 656844. [Google Scholar] [CrossRef] [PubMed]
- Jeters, R.T.; Rivera, A.J.; Boucek, L.M.; Stumpf, R.M.; Leigh, S.R.; Salyers, A.A. Antibiotic resistance genes in the vaginal microbiota of primates not normally exposed to antibiotics. Microb. Drug Resist. 2009, 15, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirichoat, A.; Flórez, A.B.; Vázquez, L.; Buppasiri, P.; Panya, M.; Lulitanond, V.; Mayo, B. Antibiotic Susceptibility Profiles of Lactic Acid Bacteria from the Human Vagina and Genetic Basis of Acquired Resistances. Int. J. Mol. Sci. 2020, 21, 2594. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.C. Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 2005, 245, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C. Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol. Lett. 2008, 282, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, L.J.; Anjum, M.F.; Roberts, A.P. Detection of a Novel, and Likely Ancestral, Tn916-Like Element from a Human Saliva Metagenomic Library. Genes 2020, 11, 548. [Google Scholar] [CrossRef]
- Milanović, V.; Osimani, A.; Aquilanti, L.; Tavoletti, S.; Garofalo, C.; Polverigiani, S.; Litta-Mulondo, A.; Cocolin, L.; Ferrocino, I.; Di Cagno, R.; et al. Occurrence of antibiotic resistance genes in the fecal DNA of healthy omnivores, ovo-lacto vegetarians and vegans. Mol. Nutr. Food Res. 2017, 61, 1601098. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, X.; Qin, J.; Lu, N.; Cheng, G.; Wu, N.; Pan, Y.; Li, J.; Zhu, L.; Wang, X.; et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun. 2013, 4, 2151. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, C.; Vignaroli, C.; Zandri, G.; Aquilanti, L.; Bordoni, D.; Osimani, A.; Clementi, F.; Biavasco, F. Direct detection of antibiotic resistance genes in specimens of chicken and pork meat. Int. J. Food Microbiol. 2007, 113, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Alicea-Serrano, A.M.; Contreras, M.; Magris, M.; Hidalgo, G.; Dominguez-Bello, M.G. Tetracycline resistance genes acquired at birth. Arch. Microbiol. 2013, 195, 447–451. [Google Scholar] [CrossRef]
- Roachford, O.S.E.; Alleyne, A.T.; Kuelbs, C.; Torralba, M.G.; Nelson, K.E. The cervicovaginal microbiome and its resistome in a random selection of Afro-Caribbean women. Hum. Microbiome J. 2021, 20, 100079. [Google Scholar] [CrossRef]
- Di Simone, N.; Santamaria Ortiz, A.; Specchia, M.; Tersigni, C.; Villa, P.; Gasbarrini, A.; Scambia, G.; D’Ippolito, S. Recent Insights on the Maternal Microbiota: Impact on Pregnancy Outcomes. Front. Immunol. 2020, 11, 528202. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.; Eichelberger, K. Maternal microbiome and pregnancy outcomes. Fertil. Steril. 2015, 104, 1358–1363. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.C.; Butler, É.M.; Grigg, C.P.; Derraik, J.G.B.; Chiavaroli, V.; Walker, N.; Thampi, S.; Creagh, C.; Reynolds, A.J.; Vatanen, T.; et al. Oral administration of maternal vaginal microbes at birth to restore gut microbiome development in infants born by caesarean section: A pilot randomised placebo-controlled trial. EBioMedicine 2021, 69, 103443. [Google Scholar] [CrossRef] [PubMed]
- Nugent, R.P.; Krohn, M.A.; Hillier, S.L. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 1991, 29, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Card, R.M.; Warburton, P.J.; MacLaren, N.; Mullany, P.; Allan, E.; Anjum, M.F. Application of microarray and functional-based screening methods for the detection of antimicrobial resistance genes in the microbiomes of healthy humans. PLoS ONE 2014, 9, e86428. [Google Scholar] [CrossRef] [Green Version]
- Petricevic, L.; Kaufmann, U.; Domig, K.J.; Kraler, M.; Marschalek, J.; Kneifel, W.; Kiss, H. Rectal Lactobacillus species and their influence on the vaginal microflora: A model of male-to-female transsexual women. J. Sex. Med. 2014, 11, 2738–2743. [Google Scholar] [CrossRef]
- Lee, C.Y.; Cheu, R.K.; Lemke, M.M.; Gustin, A.T.; France, M.T.; Hampel, B.; Thurman, A.R.; Doncel, G.F.; Ravel, J.; Klatt, N.R.; et al. Quantitative modeling predicts mechanistic links between pre-treatment microbiome composition and metronidazole efficacy in bacterial vaginosis. Nat. Commun. 2020, 11, 6147. [Google Scholar] [CrossRef]
- Laghi, L.; Zagonari, S.; Patuelli, G.; Zhu, C.; Foschi, C.; Morselli, S.; Pedna, M.F.; Sambri, V.; Marangoni, A. Vaginal metabolic profiles during pregnancy: Changes between first and second trimester. PLoS ONE 2021, 16, e0249925. [Google Scholar] [CrossRef] [PubMed]
- Kroon, S.J.; Ravel, J.; Huston, W.M. Cervicovaginal microbiota, women’s health, and reproductive outcomes. Fertil. Steril. 2018, 11, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Gueimonde, M.; Salminen, S.; Isolauri, E. Presence of specific antibiotic (tet) resistance genes in infant faecal microbiota. FEMS Immunol. Med. Microbiol. 2006, 48, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Chen, H.; Wang, Q.; Li, B.; Hu, Y.; Zhao, C.; Hu, Y.; Yin, Y. Literature-Based Phenotype Survey and In Silico Genotype Investigation of Antibiotic Resistance in the Genus Bifidobacterium. Curr. Microbiol. 2020, 77, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Ferrario, C.; Viappiani, A.; Mancabelli, L.; Mangifesta, M.; Taminiau, B.; Delcenserie, V.; et al. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl. Environ. Microbiol. 2014, 80, 6383–6394. [Google Scholar] [CrossRef] [Green Version]
- Si, J.; You, H.J.; Yu, J.; Sung, J.; Ko, G. Prevotella as a Hub for Vaginal Microbiota under the Influence of Host Genetics and Their Association with Obesity. Cell Host Microbe 2017, 21, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Patterson, A.J.; Colangeli, R.; Spigaglia, P.; Scott, K.P. Distribution of specific tetracycline and erythromycin resistance genes in environmental samples assessed by macroarray detection. Environ. Microbiol. 2007, 9, 703–715. [Google Scholar] [CrossRef]
- Murphy, A.; Barich, D.; Fennessy, M.S.; Slonczewski, J.L. An Ohio State Scenic River Shows Elevated Antibiotic Resistance Genes, Including Acinetobacter Tetracycline and Macrolide Resistance, Downstream of Wastewater Treatment Plant Effluent. Microbiol. Spectr. 2021, 1, e0094121. [Google Scholar] [CrossRef]
- Donders, G.; Bellen, G.; Rezeberga, D. Aerobic vaginitis in pregnancy. BJOG 2011, 118, 1163–1170. [Google Scholar] [CrossRef]
- Yano, J.; Sobel, J.D.; Nyirjesy, P.; Sobel, R.; Williams, V.L.; Yu, Q.; Noverr, M.C.; Fidel, P.L., Jr. Current patient perspectives of vulvovaginal candidiasis: Incidence, symptoms, management and post-treatment outcomes. BMC Womens Health 2019, 19, 48. [Google Scholar] [CrossRef] [Green Version]
- Zozaya-Hinchliffe, M.; Lillis, R.; Martin, D.H.; Ferris, M.J. Quantitative PCR assessments of bacterial species in women with and without bacterial vaginosis. J. Clin. Microbiol. 2010, 48, 1812–1819. [Google Scholar] [CrossRef] [Green Version]
- Marangoni, A.; Foschi, C.; Nardini, P.; Compri, M.; Cevenini, R. Evaluation of the Versant CT/GC DNA 1.0 assay (kPCR) for the detection of extra-genital Chlamydia trachomatis and Neisseria gonorrhoeae infections. PLoS ONE 2015, 10, e0120979. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Pop, M. ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009, 37, D443–D447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for Illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Correspondence QIIME allows analysis of high throughput community sequencing data Intensity normalization improves color calling in SOLiD sequencing. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 2016, 081257. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Package “Vegan”. R Package Version 2.0–10. 2013. Available online: https://cran.r-project.org/src/contrib/Archive/vegan/vegan_2.0-10.tar.gz (accessed on 25 November 2021).
- Gupta, S.D. Point biserial correlation coefficient and its generalization. Psychometrika 1960, 25, 393–408. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | H % (n = 142) | I % (n = 51) | BV % (n = 35) | p Value |
|---|---|---|---|---|
| ermB | 49.2% (70) | 58.8% (30) | 74.2% (26) | 0.024 |
| ermF | 19.0% (27) | 29.4% (15) | 45.7% (16) | 0.003 |
| tet(M) | 71.1% (101) | 82.3% (42) | 91.4% (32) | 0.021 |
| tet(W) | 14.0% (20) | 11.7% (6) | 20.0% (7) | 0.553 |
| Gene | Phylogenetic_Name | Avg. Relative Abundance (%) | Significance a | |
|---|---|---|---|---|
| Gene Positive [+] | Gene Negative [−] | |||
| ermB | Lactobacillus | 67.1 | 76.6 | * |
| Prevotella | 5.0 | 0.8 | *** | |
| Atopobium | 2.9 | 2.6 | *** | |
| Streptococcus | 1.4 | 1.0 | * | |
| ermF | Lactobacillus | 57.2 | 76.2 | *** |
| Gardnerella | 11.6 | 9.5 | * | |
| Prevotella | 7.3 | 1.8 | *** | |
| Atopobium | 3.3 | 2.6 | *** | |
| Streptococcus | 1.9 | 1.0 | * | |
| Prevotella 6b | 1.3 | 0.1 | *** | |
| Anaerococcus | 1.1 | 0.1 | *** | |
| tet(W) | Lactobacillus | 57.4 | 73.7 | ** |
| Bifidobacterium | 4.8 | 3.1 | ** | |
| Prevotella | 7.0 | 2.5 | *** | |
| Atopobium | 1.1 | 3.0 | ** | |
| Sneathia | 1.5 | 0.6 | ** | |
| Prevotella 6 | 1.6 | 0.2 | *** | |
| Anaerococcus | 1.5 | 0.2 | ** | |
| tet(M) | Lactobacillus | 67.8 | 83.0 | *** |
| Bifidobacterium | 4.2 | 0.8 | ** | |
| Prevotella | 4.0 | 0.4 | *** | |
| Atopobium | 3.4 | 0.7 | *** | |
| Streptococcus | 1.6 | 0.0 | *** | |
| Genera | Resistance Gene | |||
|---|---|---|---|---|
| ermB | ermF | tet(W) | tet(M) | |
| Lactobacillus | −0.136 | −0.239 | −0.165 | −0.185 |
| Gardnerella | -- | 0.046 | 0.074 | -- |
| Bifidobacterium | -- | -- | 0.051 | 0.123 |
| Prevotella | 0.264 | 0.304 | 0.199 | 0.192 |
| Atopobium | 0.016 | 0.029 | −0.062 | 0.100 |
| Streptococcus | 0.029 | 0.066 | -- | 0.104 |
| Sneathia | 0.205 | -- | 0.093 | -- |
| Alloscardovia | -- | -- | -- | 0.105 |
| Ureaplasma | -- | 0.175 | -- | -- |
| Dialister | 0.154 | 0.200 | 0.153 | 0.201 |
| Prevotella 6 | 0.099 | 0.306 | 0.271 | 0.125 |
| Aerococcus | -- | 0.035 | 0.003 | 0.076 |
| Anaerococcus | 0.067 | 0.280 | 0.300 | 0.125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Severgnini, M.; Camboni, T.; Ceccarani, C.; Morselli, S.; Cantiani, A.; Zagonari, S.; Patuelli, G.; Pedna, M.F.; Sambri, V.; Foschi, C.; et al. Distribution of ermB, ermF, tet(W), and tet(M) Resistance Genes in the Vaginal Ecosystem of Women during Pregnancy and Puerperium. Pathogens 2021, 10, 1546. https://doi.org/10.3390/pathogens10121546
Severgnini M, Camboni T, Ceccarani C, Morselli S, Cantiani A, Zagonari S, Patuelli G, Pedna MF, Sambri V, Foschi C, et al. Distribution of ermB, ermF, tet(W), and tet(M) Resistance Genes in the Vaginal Ecosystem of Women during Pregnancy and Puerperium. Pathogens. 2021; 10(12):1546. https://doi.org/10.3390/pathogens10121546
Chicago/Turabian StyleSevergnini, Marco, Tania Camboni, Camilla Ceccarani, Sara Morselli, Alessia Cantiani, Sara Zagonari, Giulia Patuelli, Maria Federica Pedna, Vittorio Sambri, Claudio Foschi, and et al. 2021. "Distribution of ermB, ermF, tet(W), and tet(M) Resistance Genes in the Vaginal Ecosystem of Women during Pregnancy and Puerperium" Pathogens 10, no. 12: 1546. https://doi.org/10.3390/pathogens10121546
APA StyleSevergnini, M., Camboni, T., Ceccarani, C., Morselli, S., Cantiani, A., Zagonari, S., Patuelli, G., Pedna, M. F., Sambri, V., Foschi, C., Consolandi, C., & Marangoni, A. (2021). Distribution of ermB, ermF, tet(W), and tet(M) Resistance Genes in the Vaginal Ecosystem of Women during Pregnancy and Puerperium. Pathogens, 10(12), 1546. https://doi.org/10.3390/pathogens10121546