
Vaginal and Anal Microbiome during Chlamydia trachomatis Infections

 , , ,
, , ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Study Population and Samples Analyzed
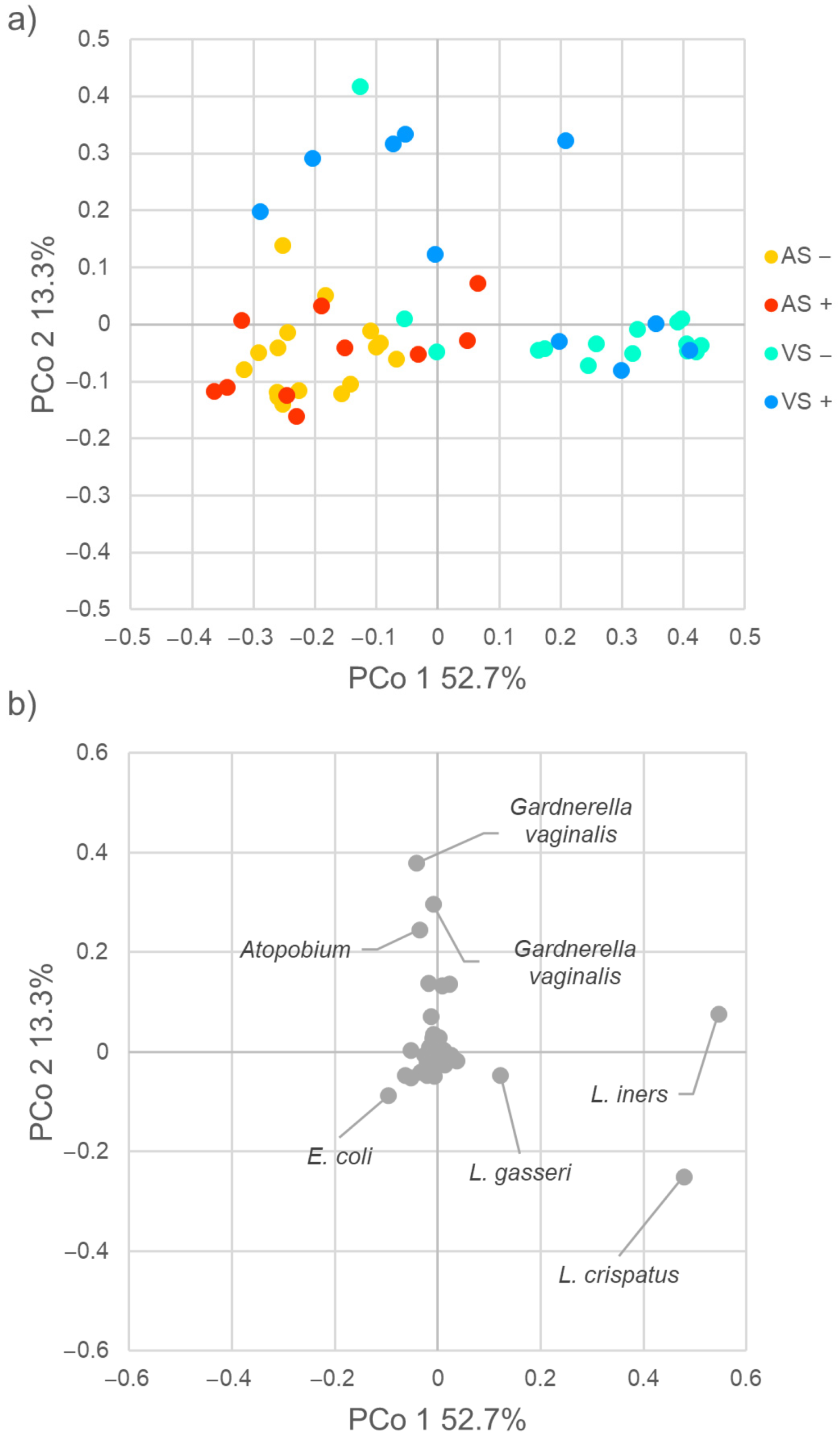
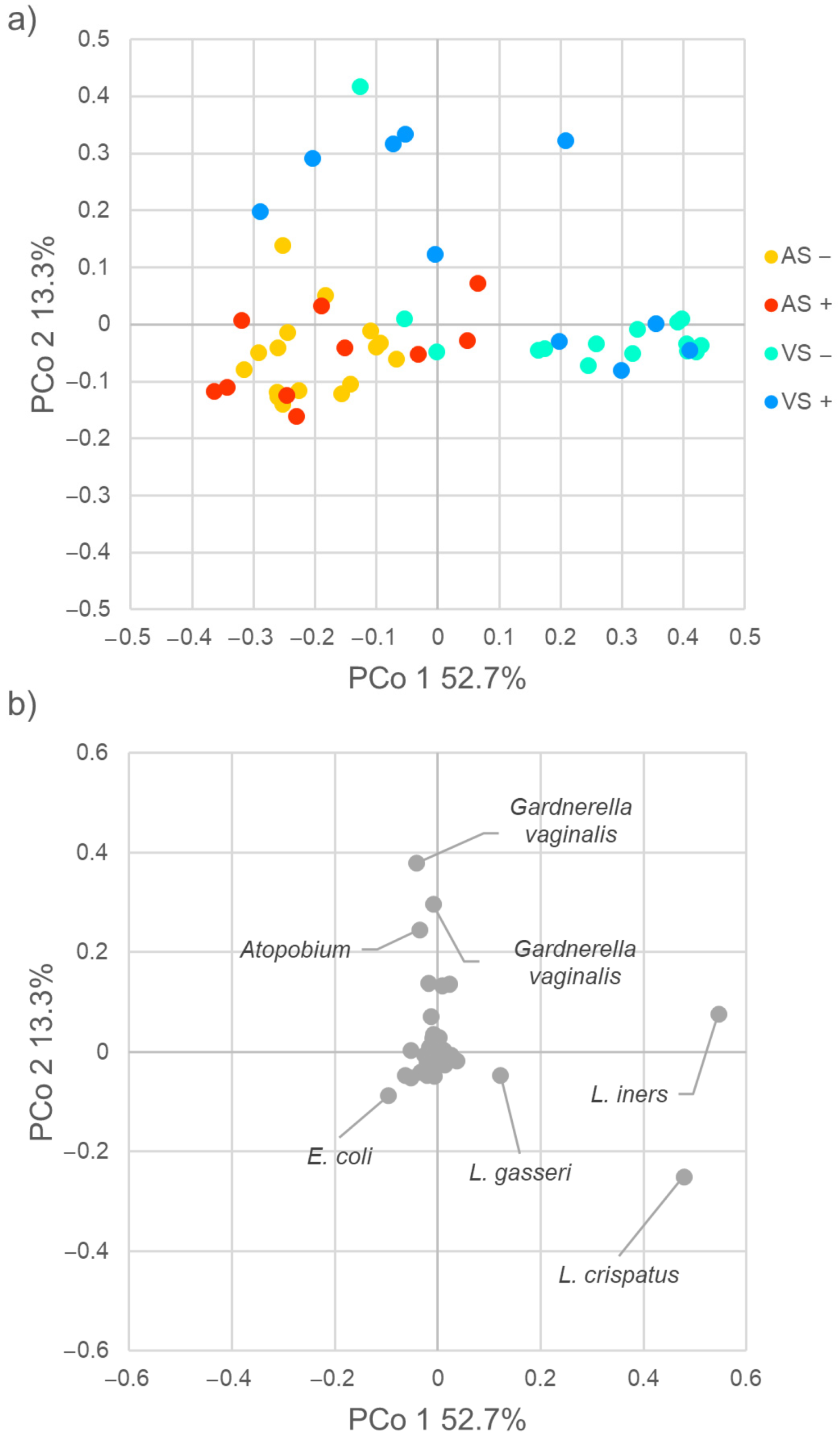
2.2. Diversity of Vaginal and Anal Microbiome
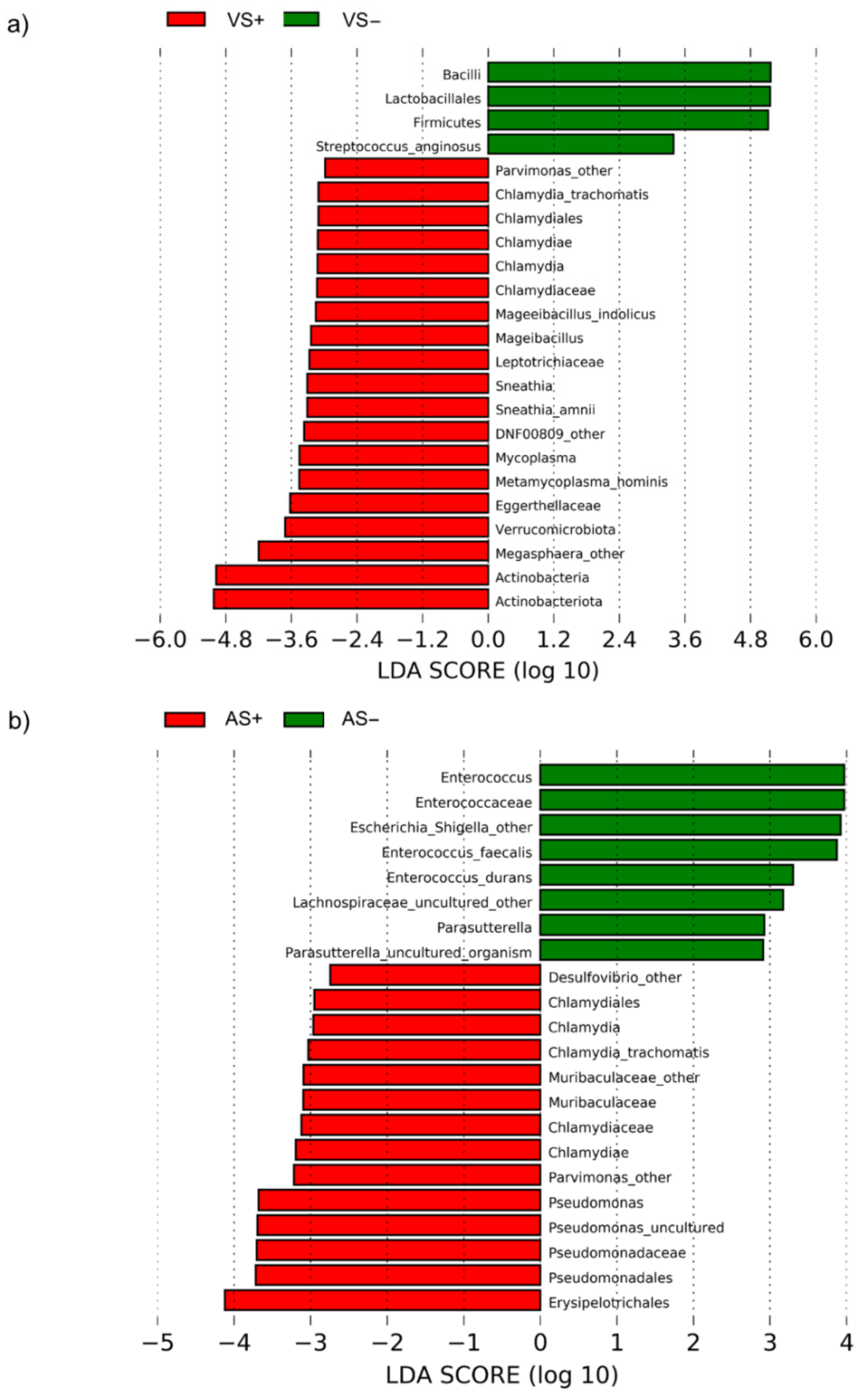
2.3. Taxonomic Composition of Vaginal and Anal Bacterial Communities
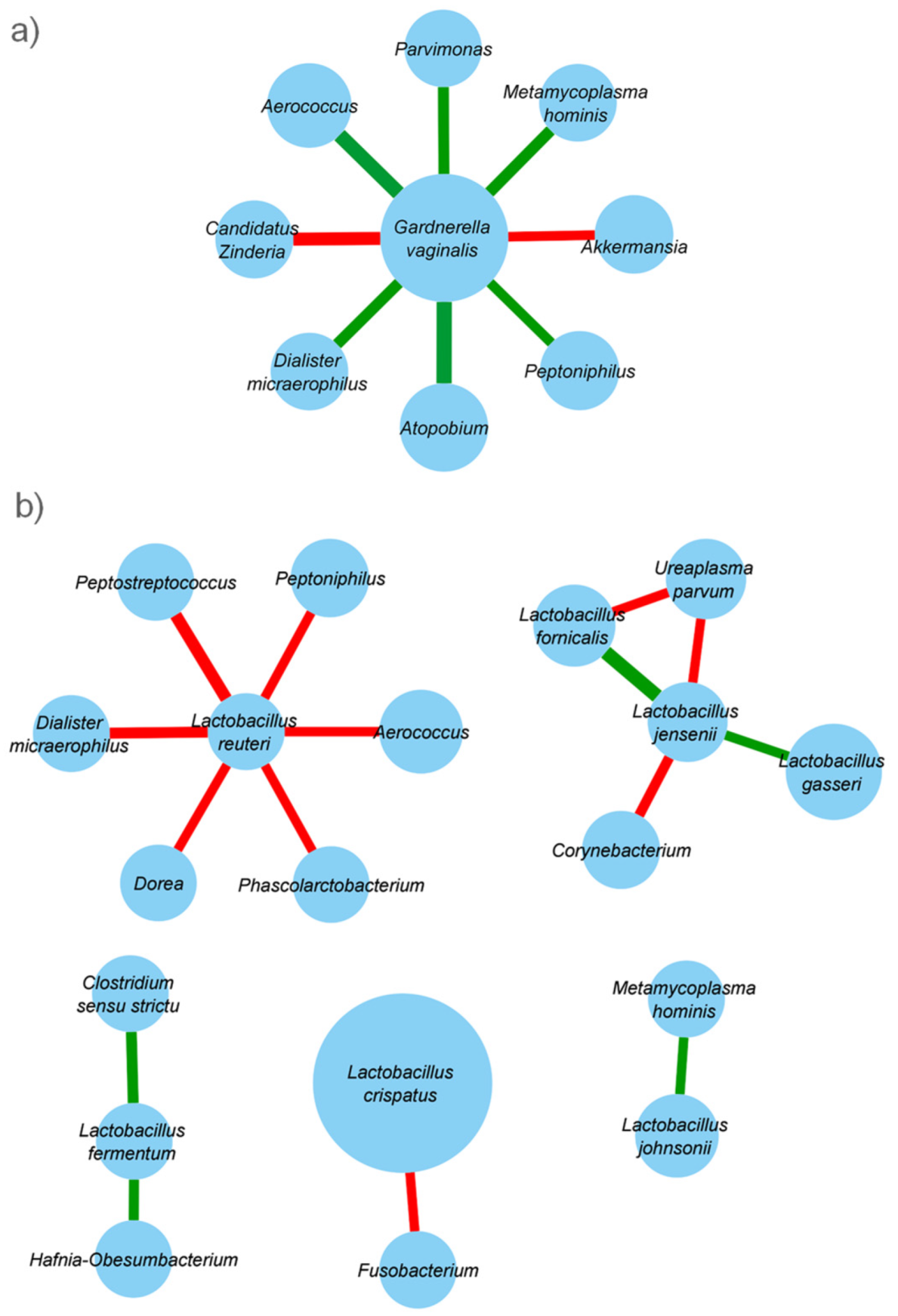
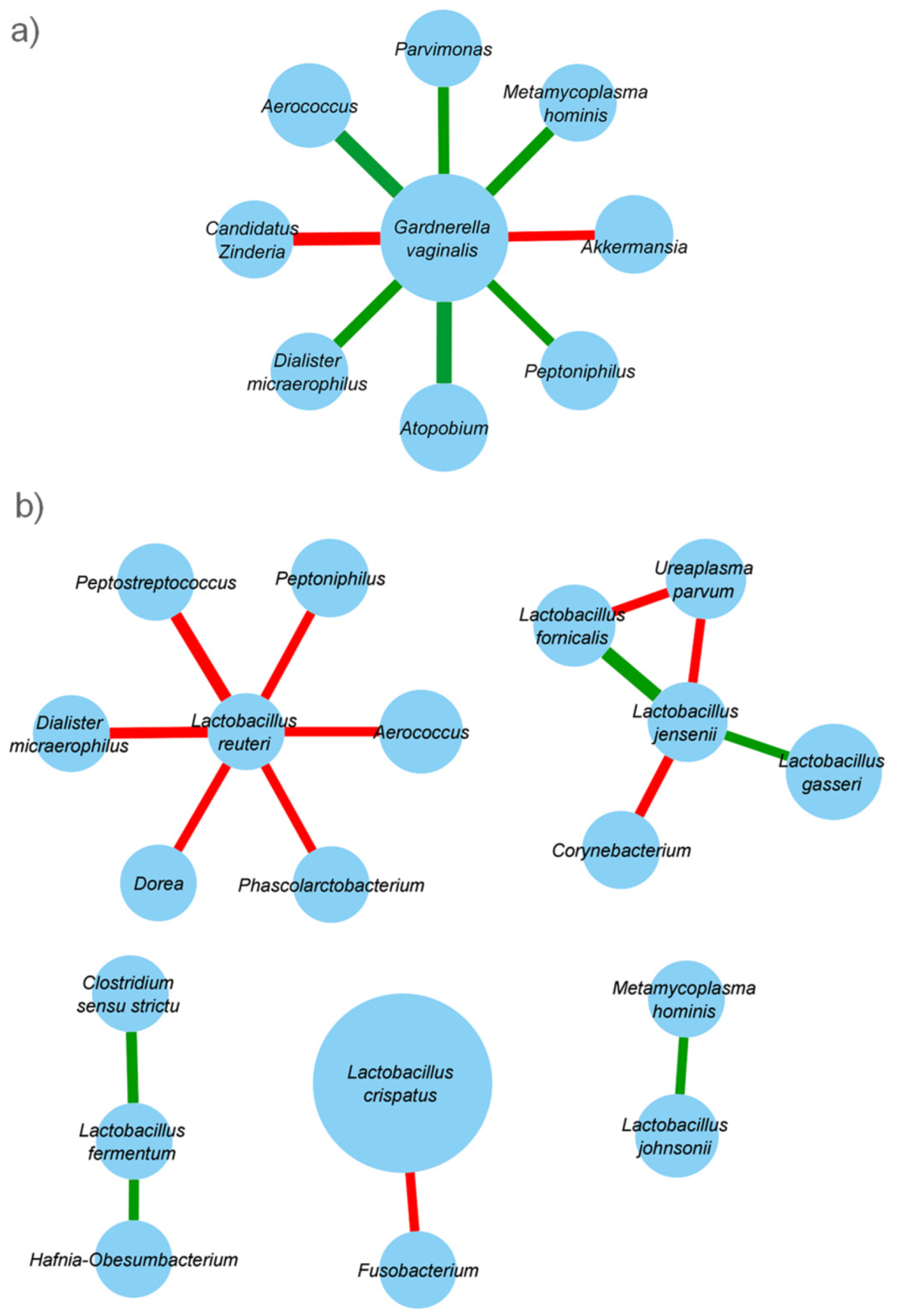
2.4. Taxonomic Co-Abundance Clusters
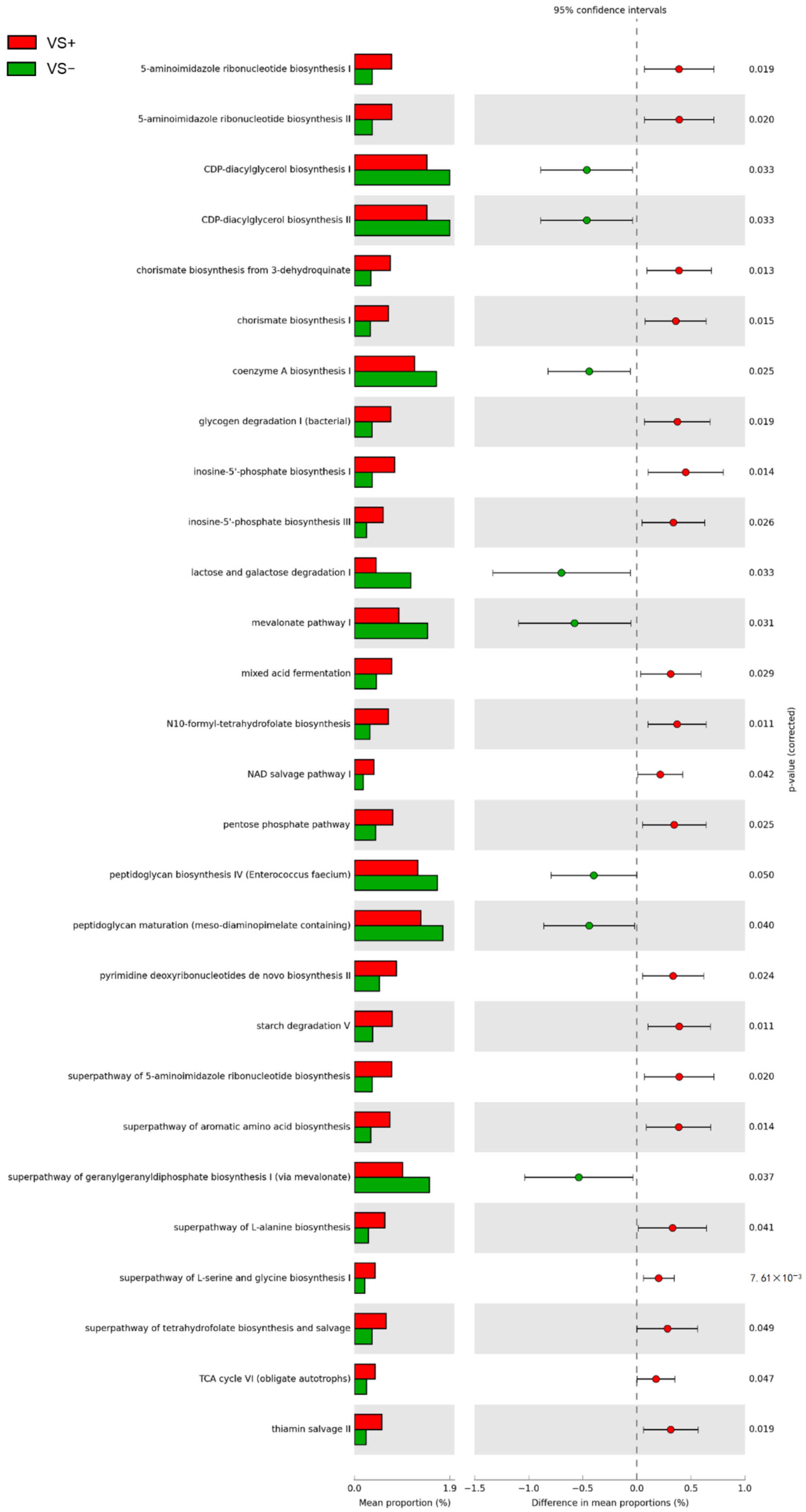
2.5. Predicted Metabolic Functions
3. Discussion
4. Materials and Methods
4.1. Study Population and Sample Collection
4.2. Diagnosis of Ano-Genital Infections
4.3. Analysis of the Vaginal and Anal Microbiome
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization (WHO). Report on Global Sexually Transmitted Infection Surveillance 2015. Available online: https://www.who.int/reproductivehealth/publications/rtis/stis-surveillance-2015/en/ (accessed on 10 September 2021).
- Price, M.J.; Ades, A.E.; De Angelis, D.; Welton, N.J.; Macleod, J.; Soldan, K.; Simms, I.; Turner, K.; Horner, P.J. Risk of pelvic inflammatory disease following Chlamydia trachomatis infection: Analysis of prospective studies with a multistate model. Am. J. Epidemiol. 2013, 178, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggerty, C.L.; Gottlieb, S.L.; Taylor, B.D.; Low, N.; Xu, F.; Ness, R.B. Risk of sequelae after Chlamydia trachomatis genital infection in women. J. Infect. Dis. 2010, 201 (Suppl. 2), S134–S155. [Google Scholar] [CrossRef] [Green Version]
- Foschi, C.; Nardini, P.; Banzola, N.; D'Antuono, A.; Compri, M.; Cevenini, R.; Marangoni, A. Chlamydia trachomatis infection prevalence and serovar distribution in a high-density urban area in the north of Italy. J. Med. Microbiol. 2016, 65, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Danby, C.S.; Cosentino, L.A.; Rabe, L.K.; Priest, C.L.; Damare, K.C.; Macio, I.S.; Meyn, L.A.; Wiesenfeld, H.C.; Hillier, S.L. Patterns of Extragenital Chlamydia and Gonorrhea in Women and Men Who Have Sex with Men Reporting a History of Receptive Anal Intercourse. Sex. Transm. Dis. 2016, 43, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.A.; Robinette, A.; Montgomery, M.; Almonte, A.; Cu-Uvin, S.; Lonks, J.R.; Chapin, K.C.; Kojic, E.M.; Hardy, E.J. Extragenital Infections Caused by Chlamydia trachomatis and Neisseria gonorrhoeae: A Review of the Literature. Infect. Dis. Obstet. Gynecol. 2016, 2016, 5758387. [Google Scholar] [CrossRef] [Green Version]
- Peters, R.P.; Dubbink, J.H.; van der Eem, L.; Verweij, S.P.; Bos, M.L.; Ouburg, S.; Lewis, D.A.; Struthers, H.; McIntyre, J.A.; Morré, S.A. Cross-sectional study of genital, rectal, and pharyngeal Chlamydia and gonorrhea in women in rural South Africa. Sex. Transm. Dis. 2014, 41, 564–569. [Google Scholar] [CrossRef] [Green Version]
- van Liere, G.A.; Hoebe, C.J.; Wolffs, P.F.; Dukers-Muijrers, N.H. High co-occurrence of anorectal chlamydia with urogenital chlamydia in women visiting an STI clinic revealed by routine universal testing in an observational study; a recommendation towards a better anorectal chlamydia control in women. BMC Infect. Dis. 2014, 14, 274. [Google Scholar] [CrossRef] [Green Version]
- van der Veer, C.; Bruisten, S.M.; van der Helm, J.J.; de Vries, H.J.; van Houdt, R. The Cervicovaginal Microbiota in Women Notified for Chlamydia trachomatis Infection: A Case-Control Study at the Sexually Transmitted Infection Outpatient Clinic in Amsterdam, The Netherlands. Clin. Infect. Dis. 2017, 64, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Tamarelle, J.; de Barbeyrac, B.; Le Hen, I.; Thiébaut, A.; Bébéar, C.; Ravel, J.; Delarocque-Astagneau, E. Vaginal microbiota composition and association with prevalent Chlamydia trachomatis infection: A cross-sectional study of young women attending a STI clinic in France. Sex. Transm. Infect. 2018, 94, 616–618. [Google Scholar] [CrossRef]
- Parolin, C.; Foschi, C.; Laghi, L.; Zhu, C.; Banzola, N.; Gaspari, V.; D’Antuono, A.; Giordani, B.; Severgnini, M.; Consolandi, C.; et al. Insights into Vaginal Bacterial Communities and Metabolic Profiles of Chlamydia trachomatis Infection: Positioning Between Eubiosis and Dysbiosis. Front. Microbiol. 2018, 9, 600. [Google Scholar] [CrossRef]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allsworth, J.E.; Peipert, J.F. Severity of bacterial vaginosis and the risk of sexually transmitted infection. Am. J. Obstet. Gynecol. 2011, 205, e1–e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Houdt, R.; Ma, B.; Bruisten, S.M.; Speksnijder, A.G.C.L.; Ravel, J.; de Vries, H.J.C. Lactobacillus iners-dominated vaginal microbiota is associated with increased susceptibility to Chlamydia trachomatis infection in Dutch women: A case-control study. Sex. Transm. Infect. 2018, 94, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Balle, C.; Lennard, K.; Dabee, S.; Barnabas, S.L.; Jaumdally, S.Z.; Gasper, M.A.; Maseko, V.; Mbulawa, Z.Z.A.; Williamson, A.L.; Bekker, L.G.; et al. Endocervical and vaginal microbiota in South African adolescents with asymptomatic Chlamydia trachomatis infection. Sci. Rep. 2018, 8, 11109. [Google Scholar] [CrossRef] [PubMed]
- Filardo, S.; Di Pietro, M.; Porpora, M.G.; Recine, N.; Farcomeni, A.; Latino, M.A.; Sessa, R. Diversity of Cervical Microbiota in Asymptomatic Chlamydia trachomatis Genital Infection: A Pilot Study. Front. Cell. Infect. Microbiol. 2017, 7, 321. [Google Scholar] [CrossRef] [PubMed]
- Ksiezarek, M.; Ugarcina-Perovic, S.; Rocha, J.; Grosso, F.; Peixe, L. Long-term stability of the urogenital microbiota of asymptomatic European women. BMC Microbiol. 2021, 21, 64. [Google Scholar] [CrossRef] [PubMed]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- Coudray, M.S.; Madhivanan, P. Bacterial vaginosis-A brief synopsis of the literature. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 245, 143–148. [Google Scholar] [CrossRef]
- Donders, G.G.G.; Bellen, G.; Grinceviciene, S.; Ruban, K.; Vieira-Baptista, P. Aerobic vaginitis: No longer a stranger. Res. Microbiol. 2017, 168, 845–858. [Google Scholar] [CrossRef]
- Borgogna, J.C.; Shardell, M.D.; Yeoman, C.J.; Ghanem, K.G.; Kadriu, H.; Ulanov, A.V.; Gaydos, C.A.; Hardick, J.; Robinson, C.K.; Bavoil, P.M.; et al. The association of Chlamydia trachomatis and Mycoplasma genitalium infection with the vaginal metabolome. Sci. Rep. 2020, 10, 3420. [Google Scholar] [CrossRef]
- Petrova, M.I.; Lievens, E.; Malik, S.; Imholz, N.; Lebeer, S. Lactobacillus species as biomarkers and agents that can promote various aspects of vaginal health. Front. Physiol. 2015, 6, 81. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.; Ravel, J.; Ma, B.; Ferreira, C.S.T.; Tristão, A.D.R.; Silva, M.G.; Marconi, C. Characteristics associated with Lactobacillus iners-dominated vaginal microbiota. Sex. Transm. Infect. 2021. online ahead of print. [Google Scholar] [CrossRef]
- France, M.T.; Ma, B.; Gajer, P.; Brown, S.; Humphrys, M.S.; Holm, J.B.; Waetjen, L.E.; Brotman, R.M.; Ravel, J. VALENCIA: A nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 2020, 8, 166. [Google Scholar] [CrossRef]
- Kelley, C.F.; Kraft, C.S.; de Man, T.J.; Duphare, C.; Lee, H.W.; Yang, J.; Easley, K.A.; Tharp, G.K.; Mulligan, M.J.; Sullivan, P.S.; et al. The rectal mucosa and condomless receptive anal intercourse in HIV-negative MSM: Implications for HIV transmission and prevention. Mucosal Immunol. 2017, 10, 996–1007. [Google Scholar] [CrossRef] [Green Version]
- Ceccarani, C.; Marangoni, A.; Severgnini, M.; Camboni, T.; Laghi, L.; Gaspari, V.; D'Antuono, A.; Foschi, C.; Re, M.C.; Consolandi, C. Rectal Microbiota Associated with Chlamydia trachomatis and Neisseria gonorrhoeae Infections in Men Having Sex with Other Men. Front. Cell. Infect. Microbiol. 2019, 9, 358. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Maldonado, J.; Kang, D.W.; Krajmalnik-Brown, R.; Caporaso, J.G. Rapidly Processed Stool Swabs Approximate Stool Microbiota Profiles. mSphere 2019, 4, e00208–e00219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budding, A.E.; Grasman, M.E.; Eck, A.; Bogaards, J.A.; Vandenbroucke-Grauls, C.M.; van Bodegraven, A.A.; Savelkoul, P.H. Rectal swabs for analysis of the intestinal microbiota. PLoS ONE 2014, 9, e101344. [Google Scholar] [CrossRef]
- Raimondi, S.; Musmeci, E.; Candeliere, F.; Amaretti, A.; Rossi, M. Identification of mucin degraders of the human gut microbiota. Sci. Rep. 2021, 11, 11094. [Google Scholar] [CrossRef]
- Petricevic, L.; Kaufmann, U.; Domig, K.J.; Kraler, M.; Marschalek, J.; Kneifel, W.; Kiss, H. Rectal Lactobacillus species and their influence on the vaginal microflora: A model of male-to-female transsexual women. J. Sex. Med. 2014, 11, 2738–2743. [Google Scholar] [CrossRef] [PubMed]
- Sobel, J.D. Vulvovaginal candidosis. Lancet 2007, 369, 1961–1971. [Google Scholar] [CrossRef]
- Foschi, C.; Zagarrigo, M.; Belletti, M.; Marangoni, A.; Re, M.C.; Gaspari, V. Genital and extra-genital Chlamydia trachomatis and Neisseria gonorrhoeae infections in young women attending a Sexually Transmitted Infections (STI) clinic. New Microbiol. 2020, 43, 115–120. [Google Scholar]
- Vasilevsky, S.; Greub, G.; Nardelli-Haefliger, D.; Baud, D. Genital Chlamydia trachomatis: Understanding the roles of innate and adaptive immunity in vaccine research. Clin. Microbiol. Rev. 2014, 27, 346–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassone-Corsi, M.; Raffatellu, M. No vacancy: How beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J. Immunol. 2015, 194, 4081–4087. [Google Scholar] [CrossRef] [Green Version]
- Castro, J.; Rosca, A.S.; Muzny, C.A.; Cerca, N. Atopobium vaginae and Prevotella bivia Are Able to Incorporate and Influence Gene Expression in a Pre-Formed Gardnerella vaginalis Biofilm. Pathogens 2021, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Diop, K.; Raoult, D.; Bretelle, F.; Fenollar, F. “Ezakiella massiliensis” sp. nov., a new bacterial species isolated from human female genital tract. New Microbes New Infect. 2016, 15, 16–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalska, K.; Wellington, S.; Maltseva, N.; Jedrzejczak, R.; Selem-Mojica, N.; RosAS−Becerra, L.R.; Barona-Gómez, F.; Hung, D.T.; Joachimiak, A. Catalytically impaired TrpA subunit of tryptophan synthase from Chlamydia trachomatis is an allosteric regulator of TrpB. Protein Sci. 2021, 30, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Sherchand, S.P.; Aiyar, A. Ammonia generation by tryptophan synthase drives a key genetic difference between genital and ocular Chlamydia trachomatis isolates. Proc. Natl. Acad Sci. USA 2019, 116, 12468–12477. [Google Scholar] [CrossRef] [Green Version]
- Watson, E.; Reid, G. Metabolomics as a clinical testing method for the diagnosis of vaginal dysbiosis. Am. J. Reprod. Immunol. 2018, 80, e12979. [Google Scholar] [CrossRef]
- Mujugira, A.; Huang, M.L.; Selke, S.; Drolette, L.; Magaret, A.S.; Wald, A. High Rate of β-Globin DNA Detection Validates Self-Sampling in Herpes Simplex Virus Shedding Studies. Sex. Transm. Dis. 2015, 42, 705–709. [Google Scholar] [CrossRef] [Green Version]
- Marangoni, A.; Foschi, C.; Nardini, P.; Compri, M.; Cevenini, R. Evaluation of the Versant CT/GC DNA 1.0 assay (kPCR) for the detection of extra-genital Chlamydia trachomatis and Neisseria gonorrhoeae infections. PLoS ONE 2015, 10, e0120979. [Google Scholar] [CrossRef]
- Foschi, C.; Gaspari, V.; Sgubbi, P.; Salvo, M.; D’Antuono, A.; Marangoni, A. Sexually transmitted rectal infections in a cohort of ‘men having sex with men’. J. Med. Microbiol. 2018, 67, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Author Correction: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–17. [Google Scholar] [CrossRef]
- Callahan, B.J. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, M.; Thurimella, K.; Lozupone, C.A. SCNIC: Sparse Correlation Network Investigation for Compositional Data. bioRxiv 2020, 11, 380733. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes–a 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raimondi, S.; Candeliere, F.; Amaretti, A.; Foschi, C.; Morselli, S.; Gaspari, V.; Rossi, M.; Marangoni, A. Vaginal and Anal Microbiome during Chlamydia trachomatis Infections. Pathogens 2021, 10, 1347. https://doi.org/10.3390/pathogens10101347
Raimondi S, Candeliere F, Amaretti A, Foschi C, Morselli S, Gaspari V, Rossi M, Marangoni A. Vaginal and Anal Microbiome during Chlamydia trachomatis Infections. Pathogens. 2021; 10(10):1347. https://doi.org/10.3390/pathogens10101347
Chicago/Turabian StyleRaimondi, Stefano, Francesco Candeliere, Alberto Amaretti, Claudio Foschi, Sara Morselli, Valeria Gaspari, Maddalena Rossi, and Antonella Marangoni. 2021. "Vaginal and Anal Microbiome during Chlamydia trachomatis Infections" Pathogens 10, no. 10: 1347. https://doi.org/10.3390/pathogens10101347
APA StyleRaimondi, S., Candeliere, F., Amaretti, A., Foschi, C., Morselli, S., Gaspari, V., Rossi, M., & Marangoni, A. (2021). Vaginal and Anal Microbiome during Chlamydia trachomatis Infections. Pathogens, 10(10), 1347. https://doi.org/10.3390/pathogens10101347