
Sickle Cell Anemia and Babesia Infection

 ,
,
Abstract
1. Introduction
2. Pathogenesis and Anemia in Babesiosis
3. Babesia and the Red Blood Cell
4. Hemoglobinopathies
5. Natural Resistance against Blood-Borne Parasites
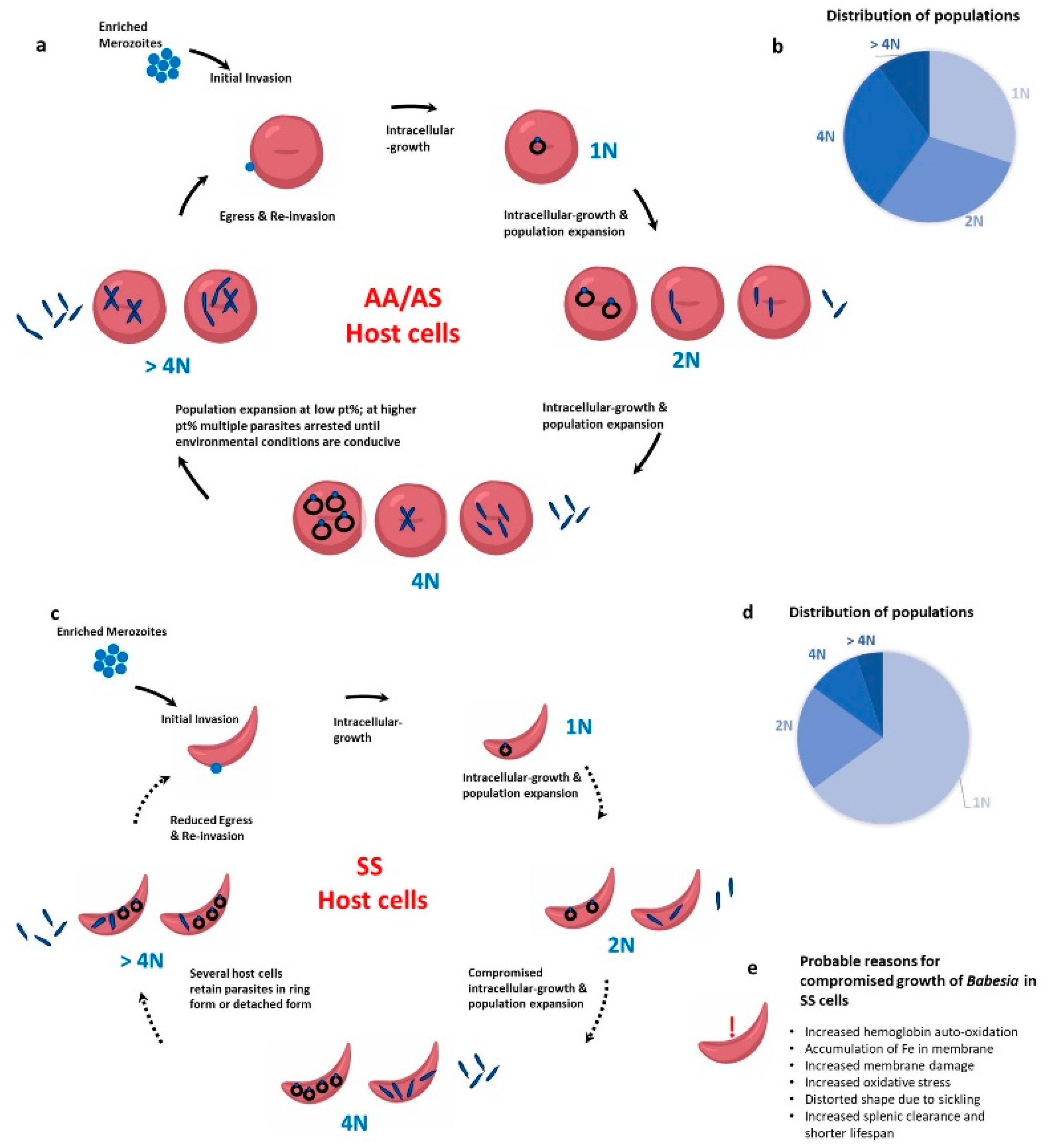
6. Babesia and the Sickle Red Cell
7. Clinical SCA and Babesiosis
8. Plausible Mechanisms for Resistance of Babesia in Sickle Cells
9. Concluding Remarks and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lobo, C.A.; Cursino-Santos, J.R.; Alhassan, A.; Rodrigues, M. Babesia: An Emerging Infectious Threat in Transfusion Medicine. PLoS Pathog. 2013, 9, e1003387. [Google Scholar] [CrossRef]
- Ord, R.L.; Lobo, C.A. Human Babesiosis: Pathogens, Prevalence, Diagnosis, and Treatment. Curr. Clin. Microbiol. Rep. 2015, 2, 173–181. [Google Scholar] [CrossRef]
- Yabsley, M.J.; Shock, B.C. Natural history of Zoonotic Babesia: Role of wildlife reservoirs. Int. J. Parasitol. Parasites Wildl. 2013, 2, 18–31. [Google Scholar] [CrossRef]
- Arrow, K.J.; Panosian, C.; Gelband, H. (Eds.) Saving Lives, Buying Time: Economics of Malaria Drugs in an Age of Resistance; National Academies Press: Washington, DC, USA, 2004. [Google Scholar]
- Lau, A.O. An overview of the Babesia, Plasmodium and Theileria genomes: A comparative perspective. Mol. Biochem. Parasitol. 2009, 164, 1–8. [Google Scholar] [CrossRef]
- Clark, I.A.; Jacobson, L.S. Do babesiosis and malaria share a common disease process? Ann. Trop. Med. Parasitol. 1998, 92, 483–488. [Google Scholar] [CrossRef]
- Raju, M.; Salazar, J.C.; Leopold, H.; Krause, P.J. Atovaquone and Azithromycin Treatment for Babesiosis in an Infant. Pediatr. Infect. Dis. J. 2007, 26, 181–183. [Google Scholar] [CrossRef]
- Slovut, D.P.; Benedetti, E.; Matas, A.J. Babesiosis and Hemophagocytic Syndrome in an Asplenic Renal Transplant Recipient. Transplantation 1996, 62, 537–539. [Google Scholar] [CrossRef]
- Brasseur, P.; Gorenflot, A. Human babesiosis in Europe. Memórias Inst. Oswaldo Cruz 1992, 87, 131–132. [Google Scholar] [CrossRef][Green Version]
- Zintl, A.; Mulcahy, G.; Skerrett, H.E.; Taylor, S.M.; Gray, J.S. Babesia divergens, a Bovine Blood Parasite of Veterinary and Zoonotic Importance. Clin. Microbiol. Rev. 2003, 16, 622–636. [Google Scholar] [CrossRef]
- Borggraefe, I.; Yuan, J.; Telford, S.R., 3rd; Menon, S.; Hunter, R.; Shah, S.; Spielman, A.; Gelfand, J.A.; Wortis, H.H.; Vannier, E. Babesia microti Primarily Invades Mature Erythrocytes in Mice. Infect. Immun. 2006, 74, 3204–3212. [Google Scholar] [CrossRef]
- Hunfeld, K.-P.; Hildebrandt, A.; Gray, J.S. Babesiosis: Recent insights into an ancient disease. Int. J. Parasitol. 2008, 38, 1219–1237. [Google Scholar] [CrossRef]
- Yi, W.; Bao, W.; Rodriguez, M.; Liu, Y.; Singh, M.; Ramlall, V.; Cursino-Santos, J.R.; Zhong, H.; Elton, C.M.; Wright, G.J.; et al. Robust adaptive immune response against Babesia microti infection marked by low parasitemia in a murine model of sickle cell disease. Blood Adv. 2018, 2, 3462–3478. [Google Scholar] [CrossRef]
- Akel, T.; Mobarakai, N. Hematologic manifestations of babesiosis. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 6. [Google Scholar] [CrossRef]
- Krause, P.J.; Gewurz, B.E.; Hill, D.; Marty, F.M.; Vannier, E.; Foppa, I.M.; Furman, R.R.; Neuhaus, E.; Skowron, G.; Gupta, S.; et al. Persistent and Relapsing Babesiosis in Immunocompromised Patients. Clin. Infect. Dis. 2008, 46, 370–376. [Google Scholar] [CrossRef]
- Lantos, P.M.; Krause, P.J. Babesiosis: Similar to Malaria but Different. Pediatr. Ann. 2002, 31, 192–197. [Google Scholar] [CrossRef]
- White, D.J.; Talarico, J.; Chang, H.G.; Birkhead, G.S.; Heimberger, T.; Morse, D.L. Human babesiosis in New York State: Review of 139 hospitalized cases and analysis of prognostic factors. Arch. Intern. Med. 1998, 158, 2149–2154. [Google Scholar] [CrossRef]
- Hatcher, J.C.; Greenberg, P.D.; Antique, J.; Jimenez-Lucho, V.E. Severe Babesiosis in Long Island: Review of 34 Cases and Their Complications. Clin. Infect. Dis. 2001, 32, 1117–1125. [Google Scholar] [CrossRef]
- Kjemtrup, A.M.; Conrad, P.A. Human babesiosis: An emerging tick-borne disease. Int. J. Parasitol. 2000, 30, 1323–1337. [Google Scholar] [CrossRef]
- Lobo, C.A.; Cursino-Santos, J.R.; Singh, M.; Rodriguez, M. Babesia divergens: A Drive to Survive. Pathogens 2019, 8, 95. [Google Scholar] [CrossRef]
- Cursino-Santos, J.R.; Singh, M.; Pham, P.; Rodriguez, M.; Lobo, C.A. Babesia divergens builds a complex population structure composed of specific ratios of infected cells to ensure a prompt response to changing environmental conditions. Cell. Microbiol. 2015, 18, 859–874. [Google Scholar] [CrossRef]
- Cursino-Santos, J.R.; Singh, M.; Pham, P.; Lobo, C.A. A novel flow cytometric application discriminates among the effects of chemical inhibitors on various phases of Babesia divergens intraerythrocytic cycle. Cytom. Part A 2017, 91, 216–231. [Google Scholar] [CrossRef]
- Cao, A.; Moi, P. Regulation of the Globin Genes. Pediatr. Res. 2002, 51, 415–421. [Google Scholar] [CrossRef][Green Version]
- Kohne, E. Hemoglobinopathies. Dtsch. Aerzteblatt Int. 2011, 108, 532–540. [Google Scholar] [CrossRef]
- Taylor, S.M.; Cerami, C.; Fairhurst, R.M. Hemoglobinopathies: Slicing the Gordian Knot of Plasmodium falciparum Malaria Pathogenesis. PLoS Pathog. 2013, 9, e1003327. [Google Scholar] [CrossRef]
- Booth, C.; Inusa, B.; Obaro, S.K. Infection in sickle cell disease: A review. Int. J. Infect. Dis. 2010, 14, e2–e12. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Prim. 2018, 4, 18010. [Google Scholar] [CrossRef]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Noubouossie, D.; Key, N.S.; Ataga, K.I. Coagulation abnormalities of sickle cell disease: Relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev. 2016, 30, 245–256. [Google Scholar] [CrossRef]
- Piel, F.B.; Patil, A.P.; Howes, R.E.; Nyangiri, O.A.; Gething, P.W.; Dewi, M.; Temperley, W.H.; Williams, T.N.; Weatherall, D.J.; Hay, S. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. Lancet 2013, 381, 142–151. [Google Scholar] [CrossRef]
- Williams, T.N.; Weatherall, D.J. World Distribution, Population Genetics, and Health Burden of the Hemoglobinopathies. Cold Spring Harb. Perspect. Med. 2012, 2, a011692. [Google Scholar] [CrossRef]
- Goheen, M.M.; Campino, S.; Cerami, C. The role of the red blood cell in host defence against falciparum malaria: An expanding repertoire of evolutionary alterations. Br. J. Haematol. 2017, 179, 543–556. [Google Scholar] [CrossRef]
- Ojodu, J.; Hulihan, M.M.; Pope, S.N.; Grant, A.M.; Centers for Disease Control and Prevention. Incidence of sickle cell trait—United States, 2010. MMWR Morb. Mortal. Wkly. Rep. 2014, 63, 1155–1158. [Google Scholar]
- Chonat, S.; Quinn, C.T. Current Standards of Care and Long Term Outcomes for Thalassemia and Sickle Cell Disease. Neurobiol. Essent. Fat. Acids 2017, 1013, 59–87. [Google Scholar] [CrossRef]
- Wiwanitkit, V. Single amino acid substitution in important hemoglobinopathies does not disturb molecular function and biological process. Int. J. Nanomed. 2008, 3, 225–227. [Google Scholar] [CrossRef][Green Version]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Luzzatto, L. Sickle Cell Anaemia and Malaria. Mediterr. J. Hematol. Infect. Dis. 2012, 4, e2012065. [Google Scholar] [CrossRef]
- Bunn, H.F. The triumph of good over evil: Protection by the sickle gene against malaria. Blood 2013, 121, 20–25. [Google Scholar] [CrossRef]
- Díaz-Castillo, A.; Contreras-Puentes, N.; Alvear-Sedán, C.; Moneriz-Pretell, C.; Rodríguez-Cavallo, E.; Mendez-Cuadro, D. Sickle Cell Trait Induces Oxidative Damage on Plasmodium falciparum Proteome at Erythrocyte Stages. Int. J. Mol. Sci. 2019, 20, 5769. [Google Scholar] [CrossRef] [PubMed]
- Senok, A.; Nelson, E.; Li, K.; Oppenheimer, S. Thalassaemia trait, red blood cell age and oxidant stress: Effects on Plasmodium falciparum growth and sensitivity to artemisinin. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 585–589. [Google Scholar] [CrossRef]
- Cyrklaff, M.; Srismith, S.; Nyboer, B.; Burda, K.; Hoffmann, A.; Lasitschka, F.; Adjalley, S.; Bisseye, C.; Simpore, J.; Mueller, A.-K.; et al. Oxidative insult can induce malaria-protective trait of sickle and fetal erythrocytes. Nat. Commun. 2016, 7, 13401. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L.; Nwachuku-Jarrett, E.; Reddy, S. Increased Sickling of Parasitised Erythrocytes as Mechanism of Resistance against Malaria in the Sickle-Cell Trait. Lancet 1970, 295, 319–322. [Google Scholar] [CrossRef]
- Roth, E.F., Jr.; Friedman, M.; Ueda, Y.; Tellez, I.; Trager, W.; Nagel, R.L. Sickling Rates of Human as Red Cells Infected in Vitro with Plasmodium falciparum Malaria. Science 1978, 202, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.J. Erythrocytic mechanism of sickle cell resistance to malaria. Proc. Natl. Acad. Sci. USA 1978, 75, 1994–1997. [Google Scholar] [CrossRef]
- Pasvol, G.; Weatherall, D.J.; Wilson, R.J.M. Cellular mechanism for the protective effect of haemoglobin S against P. falciparum malaria. Nat. Cell Biol. 1978, 274, 701–703. [Google Scholar] [CrossRef]
- Friedman, M.J. Ultrastructural Damage to the Malaria Parasite in the Sickled Cell. J. Protozool. 1979, 26, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, M.J.; Ndungu, F.; Baird, K.L.; Muller, D.P.R.; Marsh, K.; Newton, C. Oxidative stress and erythrocyte damage in Kenyan children with severe Plasmodium falciparum malaria. Br. J. Haematol. 2001, 113, 486–491. [Google Scholar] [CrossRef]
- Hebbel, R.P. Beyond hemoglobin polymerization: The red blood cell membrane and sickle disease pathophysiology. Blood 1991, 77, 214–237. [Google Scholar] [CrossRef]
- Fairhurst, R.M.; Baruch, D.I.; Brittain, N.J.; Ostera, G.R.; Wallach, J.S.; Hoang, H.L.; Hayton, K.; Guindo, A.; Makobongo, M.O.; Schwartz, O.M.; et al. Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nat. Cell Biol. 2005, 435, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Cholera, R.; Brittain, N.J.; Gillrie, M.R.; Lopera-Mesa, T.M.; Diakité, S.A.S.; Arie, T.; Krause, M.A.; Guindo, A.; Tubman, A.; Fujioka, H.; et al. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proc. Natl. Acad. Sci. USA 2008, 105, 991–996. [Google Scholar] [CrossRef]
- Santos, J.R.C.; Singh, M.; Senaldi, E.; Manwani, D.; Yazdanbakhsh, K.; Lobo, C.A. Altered parasite life-cycle processes characterize Babesia divergens infection in human sickle cell anemia. Haematologica 2019, 104, 2189–2199. [Google Scholar] [CrossRef]
- Herwaldt, B.L.; Linden, J.V.; Bosserman, E.; Young, C.; Olkowska, D.; Wilson, M. Transfusion-Associated Babesiosis in the United States: A Description of Cases. Ann. Intern. Med. 2011, 155, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.E.; Krause, P.J. Transfusion-transmitted babesiosis: Is it time to screen the blood supply? Curr. Opin. Hematol. 2016, 23, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Karkoska, K.; Louie, J.; Appiah-Kubi, A.O.; Wolfe, L.; Rubin, L.; Rajan, S.; Aygun, B. Transfusion-transmitted babesiosis leading to severe hemolysis in two patients with sickle cell anemia. Pediatr. Blood Cancer 2018, 65, e26734. [Google Scholar] [CrossRef]
- Bloch, E.M.; Herwaldt, B.L.; Leiby, D.A.; Shaieb, A.; Herron, R.M.; Chervenak, M.; Reed, W.; Hunter, R.; Ryals, R.; Hagar, W.; et al. The third described case of transfusion-transmitted Babesia duncani. Transfusion 2012, 52, 1517–1522. [Google Scholar] [CrossRef]
- Herbst, J.; Crissinger, T.; Baldwin, K. Diffuse Ischemic Strokes and Sickle Cell Crisis Induced by Disseminated Anaplasmosis: A Case Report. Case Rep. Neurol. 2019, 11, 271–276. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Hemoglobinopathy | Mutation | Position |
|---|---|---|
| HbS | Glutamic Acid to Valine | β-6 |
| HbC | Glutamic Acid to Lysine | β-6 |
| HbE | Glutamic Acid to Lysine | β-26 |
| Thalassemia | Gene Modifications | Disease Name |
| α-Thalassemia | - - / - α | HbH disease |
| - - / - - | α-Thalassemia major | |
| β-Thalassemia | β°/β | β-Thalassemia minor |
| β°/β° | β-Thalassemia major |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beri, D.; Singh, M.; Rodriguez, M.; Yazdanbakhsh, K.; Lobo, C.A. Sickle Cell Anemia and Babesia Infection. Pathogens 2021, 10, 1435. https://doi.org/10.3390/pathogens10111435
Beri D, Singh M, Rodriguez M, Yazdanbakhsh K, Lobo CA. Sickle Cell Anemia and Babesia Infection. Pathogens. 2021; 10(11):1435. https://doi.org/10.3390/pathogens10111435
Chicago/Turabian StyleBeri, Divya, Manpreet Singh, Marilis Rodriguez, Karina Yazdanbakhsh, and Cheryl Ann Lobo. 2021. "Sickle Cell Anemia and Babesia Infection" Pathogens 10, no. 11: 1435. https://doi.org/10.3390/pathogens10111435
APA StyleBeri, D., Singh, M., Rodriguez, M., Yazdanbakhsh, K., & Lobo, C. A. (2021). Sickle Cell Anemia and Babesia Infection. Pathogens, 10(11), 1435. https://doi.org/10.3390/pathogens10111435