
Viral Hepatitis and Hepatocellular Carcinoma: State of the Art

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Hepatitis A
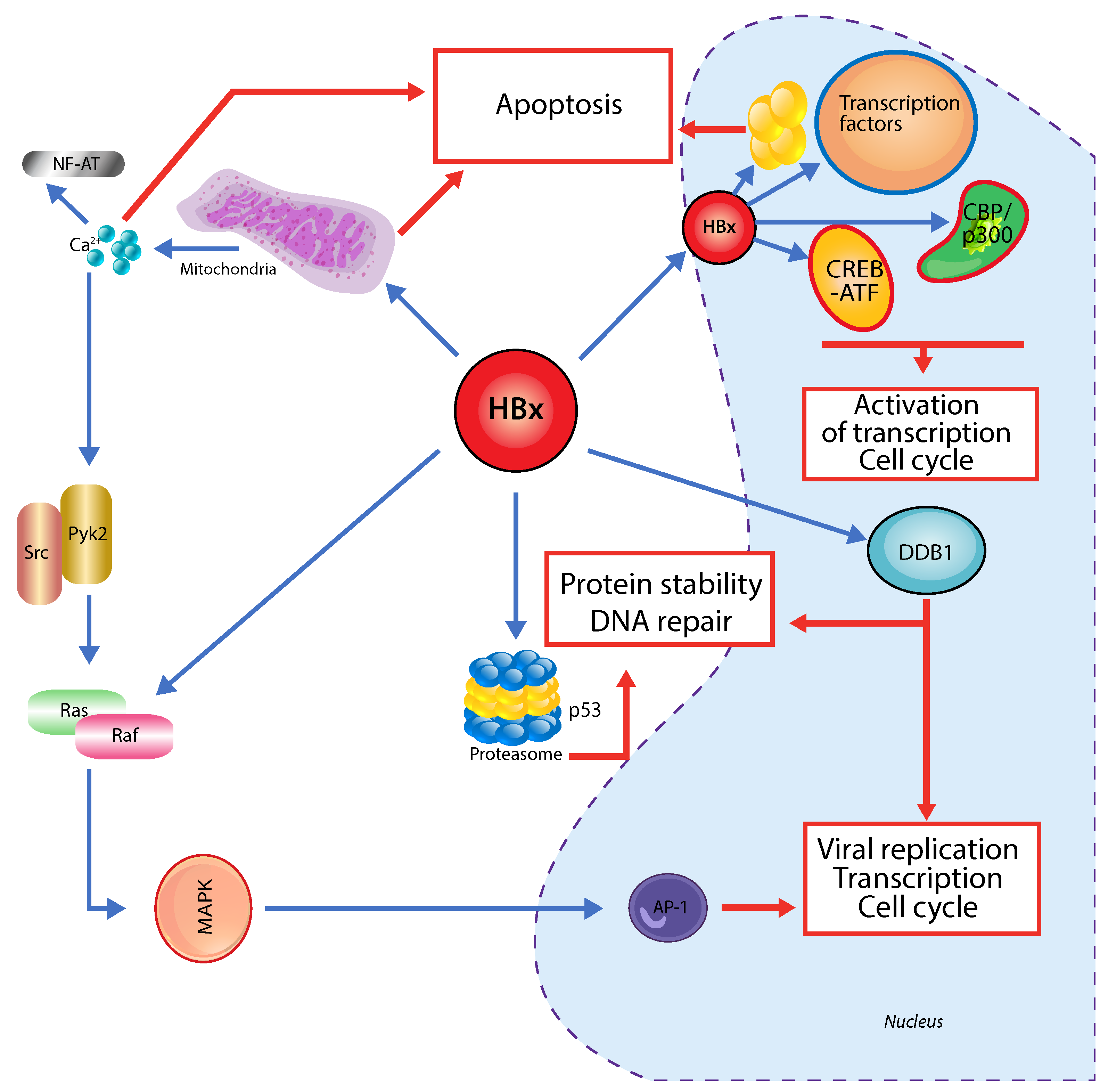
3. Hepatitis B
4. Hepatitis C
5. Hepatitis D
6. Hepatitis E
7. Discussion
8. Conclusions
9. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Singal, A.G.; El-Serag, H.B. Hepatocellular Carcinoma from Epidemiology to Prevention: Translating Knowledge into Practice. Clin. Gastroenterol. Hepatol. 2015, 13, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies from 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated with Direct-acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of Hepatocellular Carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome Sequencing of Hepatocellular Carcinomas Identifies New Mutational Signatures and Potential Therapeutic Targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated Analysis of Somatic Mutations and Focal Copy-number Changes Identifies Key Genes and Pathways in Hepatocellular Carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Abutaleb, A.; Kottilil, S. Hepatitis A: Epidemiology, Natural History, Unusual Clinical Manifestations, and Prevention. Gastroenterol. Clin. N. Am. 2020, 49, 191–199. [Google Scholar] [CrossRef]
- Jacobsen, K.H.; Koopman, J.S. Declining Hepatitis A Seroprevalence: A Global Review and Analysis. Epidemiol. Infect. 2004, 132, 1005–1022. [Google Scholar] [CrossRef]
- Aggarwal, R.; Goel, A. Hepatitis A: Epidemiology in Resource-poor Countries. Curr. Opin. Infect. Dis. 2015, 28, 488–496. [Google Scholar] [CrossRef]
- Nelson, N.P.; Weng, M.K.; Hofmeister, M.G.; Moore, K.L.; Doshani, M.; Kamili, S.; Koneru, A.; Haber, P.; Hagan, L.; Romero, J.R.; et al. Prevention of Hepatitis A Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices, 2020. MMWR Recomm. Rep. 2020, 69, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Shin, J.; Oh, J.; Jeong, K.; Choi, Y.; Park, Q.; Song, M.; Lee, D. Identification of Transfusion-transmitted Hepatitis A through Postdonation Information in Korea: Results of an HAV Lookback (2007–2012). Vox Sang. 2018, 113, 547–554. [Google Scholar] [CrossRef]
- Jacobsen, K.H. Globalization and the Changing Epidemiology of Hepatitis A Virus. Cold Spring Harb. Perspect. Med. 2018, 8, a031716. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.C.; Jeong, S.H. Natural History, Clinical Manifestations, and Pathogenesis of Hepatitis A. Cold Spring Harb. Perspect. Med. 2018, 8, a031708. [Google Scholar] [CrossRef]
- Kanda, T.; Sasaki, R.; Masuzaki, R.; Takahashi, H.; Mizutani, T.; Matsumoto, N.; Nirei, K.; Moriyama, M. Co-occurrence of Hepatitis A Infection and Chronic Liver Disease. Int. J. Mol. Sci. 2020, 21, 6384. [Google Scholar] [CrossRef] [PubMed]
- Tabor, E.; Trichopoulos, D.; Manousos, O.; Zavitsanos, X.; Drucker, J.A.; Gerety, R.J. Absence of an Association Between Past Infection with Hepatitis A Virus and Primary Hepatocellular Carcinoma. Int. J. Epidemiol. 1980, 9, 221–224. [Google Scholar] [CrossRef]
- Drucker, J.; Coursaget, P.; Maupas, P.; Goudeau, A.; Gerety, R.; Chiron, J.; Denis, F.; Mar, D. Hepatitis A Infection and Primary Hepatocellular Carcinoma. Biomedicine 1979, 31, 23–25. [Google Scholar]
- Singh, G.; Palaniappan, S.; Rotimi, O.; Hamlin, P. Autoimmune Hepatitis Triggered by Hepatitis A. Gut 2007, 56, 304. [Google Scholar] [CrossRef]
- S-Are, V.; Yoder, L.; Samala, N.; Nephew, L.; Lammert, C.; Vuppalanchi, R. An Outbreak Presents an Opportunity to Learn About A Rare Phenotype: Autoimmune Hepatitis After Acute Hepatitis A. Ann. Hepatol. 2020, 19, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, A.; Mazumder, S.; Al-Kourainy, N. The Role of Hepatocellular Carcinoma Surveillance in Autoimmune Hepatitis. Cureus 2020, 12, e11269. [Google Scholar] [CrossRef] [PubMed]
- Valean, S.; Acalovschi, M.; Dumitrascu, D.L.; Ciobanu, L.; Nagy, G.; Chira, R. Hepatocellular Carcinoma in Patients with Autoimmune Hepatitis—A Systematic Review of the Literature Published between 1989 and 2016. Med. Pharm. Rep. 2019, 92, 99–105. [Google Scholar] [CrossRef]
- Fainboim, L.; Cañero Velasco, M.C.; Marcos, C.Y.; Ciocca, M.; Roy, A.; Theiler, G.; Capucchio, M.; Nuncifora, S.; Sala, L.; Zelazko, M. Protracted, but not Acute, Hepatitis A Virus Infection is Strongly Associated with HLA-DRB1* 1301, a Marker for Pediatric Autoimmune Hepatitis. Hepatology 2001, 33, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Lohse, A.W. Autoimmune Hepatitis on the Rise. J. Hepatol. 2014, 60, 478–479. [Google Scholar] [CrossRef]
- Czaja, A.J. Acute and Acute Severe (Fulminant) Autoimmune Hepatitis. Dig. Dis. Sci. 2013, 58, 897–914. [Google Scholar] [CrossRef]
- Vento, S.; Garofano, T.; Dolci, L.; Di Perri, G.; Concia, E.; Bassetti, D. Identification of Hepatitis A Virus as a Trigger for Autoimmune Chronic Hepatitis Type 1 in Susceptible Individuals. Lancet 1991, 337, 1183–1187. [Google Scholar] [CrossRef]
- Tuohy, V.K.; Yu, M.; Yin, L.; Kawczak, J.A.; Johnson, J.M.; Mathisen, P.M.; Weinstock-Guttnnan, B.; Kinkel, R.P. The Epitope Spreading Cascade During Progression of Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Immunol. Rev. 1998, 164, 93–100. [Google Scholar] [CrossRef]
- Srinivasappa, J.; Saegusa, J.; Prabhakar, B.S.; Gentry, M.K.; Buchmeier, M.J.; Wiktor, T.J.; Koprowski, H.; Oldstone, M.; Notkins, A. Molecular Mimicry: Frequency of Reactivity of Monoclonal Antiviral Antibodies with Normal Tissues. J. Virol. 1986, 57, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Oleszak, E.L.; Lin, W.L.; Legido, A.; Melvin, J.; Hardison, H.; Hoffman, B.E.; Katsetos, C.D.; Platsoucas, C.D. Presence of Oligoclonal T Cells in Cerebrospinal Fluid of a Child with Multiphasic Disseminated Encephalomyelitis Following Hepatitis A Virus Infection. Clin. Diagn. Lab. Immunol. 2001, 8, 984–992. [Google Scholar] [CrossRef]
- Trujillo-Ochoa, J.L.; Corral-Jara, K.F.; Charles-Nino, C.L.; Panduro, A.; Fierro, N.A. Conjugated Bilirubin Upregulates TIM-3 Expression on CD4+ CD25+ T Cells: Anti-inflammatory Implications for Hepatitis a Virus Infection. Viral. Immunol. 2018, 31, 223–232. [Google Scholar] [CrossRef]
- Zhou, G.; Sprengers, D.; Boor, P.P.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; De Jonge, J.; Gaspersz, M.; et al. Antibodies against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B Virus Infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Ringehan, M.; McKeating, J.A.; Protzer, U. Viral Hepatitis and Liver Cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160274. [Google Scholar] [CrossRef]
- Parkin, D.M. The Global Health Burden of Infection-Associated Cancers in the Year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- Hassan, M.M.; Hwang, L.Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk Factors for Hepatocellular Carcinoma: Synergism of Alcohol with Viral Hepatitis and Diabetes Mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef]
- Sumi, H.; Yokosuka, O.; Seki, N.; Arai, M.; Imazeki, F.; Kurihara, T.; Kanda, T.; Fukai, K.; Kato, M.; Saisho, H. Influence of Hepatitis B Virus Genotypes on the Progression of Chronic Type B Liver Disease. Hepatology 2003, 37, 19–26. [Google Scholar] [CrossRef]
- Westland, C.; Delaney IV, W.; Yang, H.; Chen, S.S.; Marcellin, P.; Hadziyannis, S.; Gish, R.; Fry, J.; Brosgart, C.; Gibbs, C.; et al. Hepatitis B Virus Genotypes and Virologic Response in 694 Patients in Phase III Studies of Adefovir Dipivoxil. Gastroenterology 2003, 125, 107–116. [Google Scholar] [CrossRef]
- Yotsuyanagi, H.; Hino, K.; Tomita, E.; Toyoda, J.; Yasuda, K.; Iino, S. Precore and Core Promoter Mutations, Hepatitis B Virus DNA Levels and Progressive Liver Injury in Chronic Hepatitis B. J. Hepatol. 2002, 37, 355–363. [Google Scholar] [CrossRef]
- Chen, C.J.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Lu, S.N.; Huang, G.T.; Iloeje, U.H.; for the REVEAL-HBV Study Group. Risk of Hepatocellular Carcinoma across a Biological Gradient of Serum Hepatitis B Virus DNA Level. JAMA 2006, 295, 65–73. [Google Scholar] [CrossRef]
- Paterlini, P.; Driss, F.; Nalpas, B.; Pisi, E.; Franco, D.; Berthelot, P.; Bréchot, C. Persistence of Hepatitis B and Hepatitis C Viral Genomes in Primary Liver Cancers from HBsAg-Negative Patients: A Study of a Low-Endemic Area. Hepatology 1993, 17, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kobayashi, M.; Someya, T.; Saitoh, S.; Hosaka, T.; Akuta, N.; Suzuki, F.; Suzuki, Y.; Arase, Y.; Kumada, H. Occult Hepatitis B Virus Infection Increases Hepatocellular Carcinogenesis by Eight Times in Patients with Non-B, Non-C Liver Cirrhosis: A Cohort Study. J. Viral. Hepat. 2009, 16, 437–443. [Google Scholar] [CrossRef]
- Chan, H.L.; Hui, A.; Wong, M.; Tse, A.M.; Hung, L.C.; Wong, V.W.; Sung, J.J. Genotype C Hepatitis B Virus Infection Is Associated with an Increased Risk of Hepatocellular Carcinoma. Gut 2004, 53, 1494–1498. [Google Scholar] [CrossRef] [PubMed]
- Neuveut, C.; Wei, Y.; Buendia, M.A. Mechanisms of HBV-related Hepatocarcinogenesis. J. Hepatol. 2010, 52, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Arauz-Ruiz, P.; Norder, H.; Robertson, B.H.; Magnius, L.O. Genotype H: A New Amerindian Genotype of Hepatitis B Virus Revealed in Central America. J. Gen. Virol. 2002, 83, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; Couroucé, A.M.; Magnius, L.O. Complete Genomes, Phylogenetic Relatedness, and Structural Proteins of Six Strains of the Hepatitis B Virus, Four of Which Represent Two New Genotypes. Virology 1994, 198, 489–503. [Google Scholar] [CrossRef]
- Stuyver, L.; De Gendt, S.; Van Geyt, C.; Zoulim, F.; Fried, M.; Schinazi, R.F.; Rossau, R. A New Genotype of Hepatitis B Virus: Complete Genome and Phylogenetic Relatedness. J. Gen. Virol. 2000, 81, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Fujie, H.; Moriya, K.; Shintani, Y.; Yotsuyanagi, H.; Iino, S.; Kimura, S.; Koike, K. Hepatitis B Virus Genotypes and Hepatocellular Carcinoma in Japan. Gastroenterology 2001, 120, 1564–1565. [Google Scholar] [CrossRef]
- Yang, H.I.; Lu, S.N.; Liaw, Y.F.; You, S.L.; Sun, C.A.; Wang, L.Y.; Hsiao, C.K.; Chen, P.J.; Chen, D.S.; Chen, C.J. Hepatitis B e Antigen and the Risk of Hepatocellular Carcinoma. N. Engl. J. Med. 2002, 347, 168–174. [Google Scholar] [CrossRef]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Basal Core Promoter Mutations of Hepatitis B Virus Increase the Risk of Hepatocellular Carcinoma in Hepatitis B Carriers. Gastroenterology 2003, 124, 327–334. [Google Scholar] [CrossRef]
- Kuang, S.Y.; Jackson, P.E.; Wang, J.B.; Lu, P.X.; Muñoz, A.; Qian, G.S.; Kensler, T.W.; Groopman, J.D. Specific Mutations of Hepatitis B Virus in Plasma Predict Liver Cancer Development. Proc. Natl. Acad. Sci. USA 2004, 101, 3575–3580. [Google Scholar] [CrossRef]
- Yang, Z.; Zhuang, L.; Lu, Y.; Xu, Q.; Tang, B.; Chen, X. Naturally Occurring Basal Core Promoter A1762T/G1764A Dual Mutations Increase the Risk of HBV-Related Hepatocellular Carcinoma: A Meta-Analysis. Oncotarget 2016, 7, 12525–12536. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between Hepatitis B Virus Mutations and the Risk of Hepatocellular Carcinoma: A Meta-Analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Tanaka, Y.; Fong, D.Y.T.; Fung, J.; Wong, D.K.H.; Yuen, J.C.H.; However, D.Y.K.; Chan, A.O.O.; Wong, B.C.Y.; Mizokami, M.; et al. Independent Risk Factors and Predictive Score for the Development of Hepatocellular Carcinoma in Chronic Hepatitis B. J. Hepatol. 2009, 50, 80–88. [Google Scholar] [CrossRef]
- Abramovitch, R.; Tavor, E.; Jacob-Hirsch, J.; Zeira, E.; Amariglio, N.; Pappo, O.; Rechavi, G.; Galun, E.; Honigman, A. A Pivotal Role of Cyclic AMP-responsive Element Binding Protein in Tumor Progression. Cancer Res. 2004, 64, 1338–1346. [Google Scholar] [CrossRef]
- Kim, Y.C.; Song, K.S.; Yoon, G.; Nam, M.J.; Ryu, W.S. Activated RAS Oncogene Collaborates with HBx Gene of Hepatitis B Virus to Transform Cells by Suppressing HBx-mediated Apoptosis. Oncogene 2001, 20, 16–23. [Google Scholar] [CrossRef]
- Di Bisceglie, A.M. Hepatitis B and Hepatocellular Carcinoma. Hepatology 2009, 49, S56–S60. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, W.; Yan, H.X.; Luo, T.; Zhang, J.; Tang, L.; Wu, F.Q.; Zhang, H.L.; Yu, L.X.; Zheng, L.Y.; et al. Hepatitis B Virus X (HBx) Induces Tumorigenicity of Hepatic Progenitor Cells in 3,5-Diethoxycarbonyl-1,4-Dihydrocollidine-Treated HBx Transgenic Mice. Hepatology 2012, 55, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.; Kramvis, A.; Kew, M.C. High Prevalence of 1762T 1764A Mutations in the Basic Core Promoter of Hepatitis B Virus Isolated from Black Africans with Hepatocellular Carcinoma Compared with Asymptomatic Carriers. Hepatology 1999, 29, 946–953. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, K.H.; Lee, J.M.; Park, J.H.; Kim, H.S. Impact of Hepatitis B Virus (HBV) X Gene Mutations on Hepatocellular Carcinoma Development in Chronic HBV Infection. Clin. Vaccine Immunol. 2011, 18, 914–921. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Zhang, T.; Ye, L.; Wang, W.; Zhang, X. Analysis of Hepatitis B Virus X Gene (HBx) Mutants in Tissues of Patients Suffered from Hepatocellular Carcinoma in China. Cancer Epidemiol. 2012, 36, 369–374. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, J.; Wang, Y.; Wang, A.; Guo, H.; Wei, F.; Mehta, S.R.; Espitia, S.; Smith, D.M.; Liu, L.; et al. A Novel Mutant 10Ala/Arg Together with Mutant 144Ser/Arg of Hepatitis B Virus X Protein Involved in Hepatitis B Virus-related Hepatocarcinogenesis in HepG2 Cell Lines. Cancer Lett. 2016, 371, 285–291. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and Viral Genetic Variation in HBV-related Hepatocellular Carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Wungu, C.D.K.; Ariyanto, F.C.; Prabowo, G.I.; Soetjipto, S.; Handajani, R. Meta-Analysis: Association between Hepatitis B Virus Pres Mutation and Hepatocellular Carcinoma Risk. J. Viral. Hepat. 2021, 28, 61–71. [Google Scholar] [CrossRef]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cacciapuoti, C. Hepatitis C Virus (HCV) Genotypes Distribution: An Epidemiological Up-date in Europe. Infect. Agent. Cancer 2016, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Chigbu, D.I.; Loonawat, R.; Sehgal, M.; Patel, D.; Jain, P. Hepatitis C Virus Infection: Host–Virus Interaction and Mechanisms of Viral Persistence. Cells 2019, 8, 376. [Google Scholar] [CrossRef]
- Douam, F.; Lavillette, D.; Cosset, F.L. The Mechanism of HCV Entry into Host Cells. Prog. Mol. Biol. Transl. Sci. 2015, 129, 63–107. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Schulze-Osthoff, K. Apoptosis in Hepatitis C Virus Infection. Cell Death Differ. 2003, 10, S48–S58. [Google Scholar] [CrossRef]
- Irshad, M.; Gupta, P.; Irshad, K. Immunopathogenesis of Liver Injury during Hepatitis C Virus Infection. Viral Immunol. 2019, 32, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed]
- Schietroma, I.; Scheri, G.C.; Pinacchio, C.; Statzu, M.; Petruzziello, A.; Vullo, V. Hepatitis C Virus and Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 16–25. [Google Scholar] [CrossRef]
- de Oliveria Andrade, L.J.; D’Oliveira, A.; Junior, R.C.M.; De Souza, E.C.; Silva, C.A.C.; Parana, R. Association between Hepatitis C and Hepatocellular Carcinoma. J. Glob. Infect. Dis. 2009, 1, 33–37. [Google Scholar] [CrossRef]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 2631. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Shi, Y.; Zhang, M.; Goswami, S.; Afridi, S.; Meng, L.; Ma, J.; Chen, Y.; Lin, Y.; Zhang, J.; et al. Global Immune Characterization of HBV/HCV-Related Hepatocellular Carcinoma Identifies Macrophage and T-Cell Subsets Associated with Disease Progression. Cell Discov. 2020, 6, 90–105. [Google Scholar] [CrossRef]
- Hong, G.Q.; Cai, D.; Gong, J.P.; Lai, X. Innate Immune Cells and Their Interaction with T Cells in Hepatocellular Carcinoma. Oncol. Lett. 2021, 21, 57. [Google Scholar] [CrossRef]
- Lonardo, A.; Adinolfi, L.E.; Loria, P.; Carulli, N.; Ruggiero, G.; Day, C.P. Steatosis and Hepatitis C Virus: Mechanisms and Significance for Hepatic and Extrahepatic Disease. Gastroenterology 2004, 126, 586–597. [Google Scholar] [CrossRef]
- Miyoshi, H.; Moriya, K.; Tsutsumi, T.; Shinzawa, S.; Fujie, H.; Shintani, Y.; Fujinaga, H.; Goto, K.; Todoroki, T.; Suzuki, T.; et al. Pathogenesis of Lipid Metabolism Disorder in Hepatitis C: Polyunsaturated Fatty Acids Counteract Lipid Alterations Induced by the Core Protein. J. Hepatol. 2011, 54, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Loizides-Mangold, U.; Clément, S.; Alfonso-Garcia, A.; Branche, E.; Conzelmann, S.; Parisot, C.; Potma, E.O.; Riezman, H.; Negro, F. HCV 3a Core Protein Increases Lipid Droplet Cholesteryl Ester Content via a Mechanism Dependent on Sphingolipid Biosynthesis. PLoS ONE 2014, 9, e115309. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty Acid Synthase Is up-Regulated during Hepatitis C Virus Infection and Regulates Hepatitis C Virus Entry and Production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of Endoplasmic Reticulum Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Machida, K. NANOG-Dependent Metabolic Reprogramming and Symmetric Division in Tumor-Initiating Stem-like Cells. Adv. Exp. Med. Biol. 2018, 1032, 105–113. [Google Scholar] [CrossRef]
- Zhou, J.J.; Chen, R.F.; Deng, X.G.; Zhou, Y.; Ye, X.; Yu, M.; Tang, J.; He, X.Y.; Cheng, D.; Zeng, B.; et al. Hepatitis C Virus Core Protein Regulates NANOG Expression via the STAT3 Pathway. FEBS Lett. 2014, 588, 566–573. [Google Scholar] [CrossRef]
- Kumar, D.B.U.; Chen, C.L.; Liu, J.C.; Feldman, D.E.; Sher, L.S.; French, S.; DiNorcia, J.; French, S.W.; Naini, B.V.; Junrungsee, S.; et al. TLR4 Signaling via NANOG Cooperates with STAT3 to Activate Twist1 and Promote Formation of Tumor-Initiating Stem-like Cells in Livers of Mice. Gastroenterology 2016, 150, 707–719. [Google Scholar] [CrossRef]
- Chen, C.L.; Tsukamoto, H.; Machida, K. Oncogenic Signaling Pathways and Origins of Tumor-Initiating Stem-like Cells of Hepatocellular Carcinomas Induced by Hepatitis C Virus, Alcohol and/or Obesity. Hepatol. Int. 2014, 8, 330–338. [Google Scholar] [CrossRef]
- Saha, B.; Szabo, G. Innate Immune Cell Networking in Hepatitis C Virus Infection. J. Leukoc. Biol. 2014, 96, 757–766. [Google Scholar] [CrossRef]
- Rehermann, B. Hepatitis C Virus versus Innate and Adaptive Immune Responses: A Tale of Coevolution and Coexistence. J. Clin. Investig. 2009, 119, 1745–1754. [Google Scholar] [CrossRef]
- Liu, H.M.; Gale, M. Hepatitis C Virus Evasion from RIG-I-Dependent Hepatic Innate Immunity. Gastroenterol. Res. Pract. 2010, 2010, 548390. [Google Scholar] [CrossRef]
- Sumida, K.; Shimoda, S.; Iwasaka, S.; Hisamoto, S.; Kawanaka, H.; Akahoshi, T.; Ikegami, T.; Shirabe, K.; Shimono, N.; Maehara, Y.; et al. Characteristics of Splenic CD 8+ T Cell Exhaustion in Patients with Hepatitis C. Clin. Exp. Immunol. 2013, 174, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Dolina, J.S.; Braciale, T.J.; Hahn, Y.S. Liver-Primed CD8+ T Cells Suppress Antiviral Adaptive Immunity through Galectin-9-Independent T-Cell Immunoglobulin and Mucin 3 Engagement of High-Mobility Group Box 1 in Mice. Hepatology 2014, 59, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Rangachari, M.; Kuchroo, V.K. Tim-3: A Co-receptor with Diverse Roles in T Cell Exhaustion and Tolerance. Semin. Immunol. 2019, 42, 101302. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Yoon, Y.S.; Le Lay, J.; Kaestner, K.H.; Hedrick, S.; Montminy, M. CREB Pathway Links PGE2 Signaling with Macrophage Polarization. Proc. Natl. Acad. Sci. USA 2015, 112, 15642–15647. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef]
- Ahmed, F.; Ibrahim, A.; Cooper, C.L.; Kumar, A.; Crawley, A.M. Chronic Hepatitis C Virus Infection Impairs M1 Macrophage Differentiation and Contributes to CD8+ T-Cell Dysfunction. Cells 2019, 8, 374. [Google Scholar] [CrossRef]
- Ng, T.; Britton, G.J.; Hill, E.V.; Verhagen, J.; Burton, B.R.; Wraith, D.C. Regulation of Adaptive Immunity; The Role of Interleukin-10. Front. Immunol. 2013, 4, 129. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Swann, J.; Kelly, J.M.; Cretney, E.; Yokoyama, W.M.; Diefenbach, A.; Sayers, T.J.; Hayakawa, Y. NKG2D Recognition and Perforin Effector Function Mediate Effective Cytokine Immunotherapy of Cancer. J. Exp. Med. 2004, 200, 1325–1335. [Google Scholar] [CrossRef]
- Mantovani, S.; Oliviero, B.; Varchetta, S.; Mele, D.; Mondelli, M.U. Natural Killer Cell Responses in Hepatocellular Carcinoma: Implications for Novel Immunotherapeutic Approaches. Cancers 2020, 12, 926. [Google Scholar] [CrossRef]
- Strunz, B.; Hengst, J.; Deterding, K.; Manns, M.P.; Cornberg, M.; Ljunggren, H.G.; Wedemeyer, H.; Björkström, N.K. Chronic Hepatitis C Virus Infection Irreversibly Impacts Human Natural Killer Cell Repertoire Diversity. Nat. Commun. 2018, 9, 2275. [Google Scholar] [CrossRef] [PubMed]
- Ragab, D.; Samaha, D.; Mohamed, N.; Rafik, M.; Abdel Hady, W. Chronic Hepatitis C Virus Infection Impairs Natural Killer Cells–Dendritic Cells Cross-Talk: An in Vitro Culture Study. Microbiol. Immunol. 2021, 65, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Perpiñán, E.; Pérez-Del-Pulgar, S.; Londoño, M.C.; Mariño, Z.; Bartres, C.; González, P.; García-López, M.; Pose, E.; Lens, S.; Maini, M.K.; et al. Cirrhosis Hampers Early and Rapid Normalization of Natural Killer Cell Phenotype and Function in Hepatitis C Patients Undergoing Interferon-Free Therapy. Front. Immunol. 2020, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Njiomegnie, G.F.; Read, S.A.; Fewings, N.; George, J.; McKay, F.; Ahlenstiel, G. Immunomodulation of the Natural Killer Cell Phenotype and Response during HCV Infection. J. Clin. Med. 2020, 9, 1030. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+ CD25+ Regulatory T Cells Inhibit Natural Killer Cell Functions in a Transforming Growth Factor-β-Dependent Manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the mTOR Pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Zwirner, N.W.; Domaica, C.I.; Fuertes, M.B. Regulatory Functions of NK Cells during Infections and Cancer. J. Leukoc. Biol. 2021, 109, 185–194. [Google Scholar] [CrossRef]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 Fosters HCC Development by Enhancing TGF-β-Mediated Alternative Activation of Macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges. Gastroenterology 2019, 156, 431–445. [Google Scholar] [CrossRef]
- Patel, S.V.; Jayaweera, D.T.; Althoff, K.N.; Eron, J.J.; Radtchenko, J.; Mills, A.; Moyle, G.; Santiago, S.; Sax, P.E.; Gillman, J.; et al. Real-World Efficacy of Direct Acting Antiviral Therapies in Patients with HIV/HCV. PLoS ONE 2020, 15, e0228847. [Google Scholar] [CrossRef]
- Vranjkovic, A.; Deonarine, F.; Kaka, S.; Angel, J.B.; Cooper, C.L.; Crawley, A.M. Direct-Acting Antiviral Treatment of Hcv Infection Does Not Resolve the Dysfunction of Circulating CD8+ T-Cells in Advanced Liver Disease. Front. Immunol. 2019, 10, 1926. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Thimme, R. Mait Be Different–Persisting Dysfunction after DAA-Mediated Clearance of Chronic Hepatitis C Virus Infection. Eur. J. Immunol. 2016, 46, 2099–2102. [Google Scholar] [CrossRef] [PubMed]
- Langhans, B.; Nischalke, H.D.; Krämer, B.; Hausen, A.; Dold, L.; van Heteren, P.; Hüneburg, R.; Nattermann, J.; Strassburg, C.P.; Spengler, U. Increased Peripheral CD4+ Regulatory T Cells Persist after Successful Direct-Acting Antiviral Treatment of Chronic Hepatitis C. J. Hepatol. 2017, 66, 888–896. [Google Scholar] [CrossRef]
- Mehta, P.; Reddivari, A.K.R. Hepatitis; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Ferrante, N.D.; Re, V.L. Epidemiology, Natural History, and Treatment of Hepatitis Delta Virus Infection in HIV/Hepatitis B Virus Coinfection. Curr. HIV/AIDS Rep. 2020, 17, 405–414. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Manns, M.P. Epidemiology, Pathogenesis and Management of Hepatitis D: Update and Challenges Ahead. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Alfaiate, D.; Clément, S.; Gomes, D.; Goossens, N.; Negro, F. Chronic Hepatitis D and Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis of Observational Studies. J. Hepatol. 2020, 73, 533–539. [Google Scholar] [CrossRef]
- Argirion, I.; Mahale, P.; Pfeiffer, R.M.; Koshiol, J.; O’Brien, T.R. Cirrhotic Controls in a Pooled Analysis of Hepatitis D and Hepatocellular Carcinoma. J. Hepatol. 2020, 73, 1583–1584. [Google Scholar] [CrossRef]
- Su, C.W.; Huang, Y.H.; Huo, T.I.; Shih, H.H.; Sheen, I.J.; Chen, S.W.; Lee, P.C.; Lee, S.D.; Wu, J.C. Genotypes and Viremia of Hepatitis B and D Viruses Are Associated with Outcomes of Chronic Hepatitis D Patients. Gastroenterology 2006, 130, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Del Ninno, E.; Rumi, M.; Russo, A.; Sangiovanni, A.; De Franchis, R.; Ronchi, G.; Colombo, M. A 28-Year Study of the Course of Hepatitis Δ Infection: A Risk Factor for Cirrhosis and Hepatocellular Carcinoma. Gastroenterology 2009, 136, 1629–1638. [Google Scholar] [CrossRef]
- Palom, A.; Rodríguez-Tajes, S.; Navascués, C.A.; García-Samaniego, J.; Riveiro-Barciela, M.; Lens, S.; Rodríguez, M.; Esteban, R.; Buti, M. Long-Term Clinical Outcomes in Patients with Chronic Hepatitis Delta: The Role of Persistent Viraemia. Aliment. Pharmacol. Ther. 2020, 51, 158–166. [Google Scholar] [CrossRef]
- Hernàndez-Èvole, H.; Briz-Redón, Á.; Berenguer, M. Changing Delta Hepatitis Patient Profile: A Single Center Experience in Valencia Region, Spain. World J. Hepatol. 2020, 12, 277–287. [Google Scholar] [CrossRef]
- Mahale, P.; Aka, P.; Chen, X.; Pfeiffer, R.M.; Liu, P.; Groover, S.; Mendy, M.; Njie, R.; Goedert, J.J.; Kirk, G.D.; et al. Hepatitis D Virus Infection, Cirrhosis and Hepatocellular Carcinoma in the Gambia. J. Viral. Hepat. 2019, 26, 738–749. [Google Scholar] [CrossRef]
- Puigvehí, M.; Moctezuma-Velázquez, C.; Villanueva, A.; Llovet, J.M. The Oncogenic Role of Hepatitis Delta Virus in Hepatocellular Carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef] [PubMed]
- D’souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular Mechanisms of Viral Hepatitis Induced Hepatocellular Carcinoma. World J. Gastroenterol. 2020, 26, 5759. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Jeong, S.H.; Hwang, S.B. Large Hepatitis Delta Antigen Modulates Transforming Growth Factor-β Signaling Cascades: Implication of Hepatitis Delta Virus–Induced Liver Fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef]
- Majumdar, A.; Curley, S.A.; Wu, X.; Brown, P.; Hwang, J.P.; Shetty, K.; Yao, Z.X.; He, A.R.; Li, S.; Katz, L.; et al. Hepatic Stem Cells and Transforming Growth Factor β In Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 530–538. [Google Scholar] [CrossRef]
- Choi, S.S.; Diehl, A.M. Epithelial-to-Mesenchymal Transitions in the Liver. Hepatology 2009, 50, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.; Brichler, S.; Khan, E.; Chami, M.; Dény, P.; Kremsdorf, D.; Gordien, E. Large Hepatitis Delta Antigen Activates STAT-3 and NF-κB via Oxidative Stress. J. Viral. Hepat. 2012, 19, 744–753. [Google Scholar] [CrossRef]
- Chen, M.; Du, D.; Zheng, W.; Liao, M.; Zhang, L.; Liang, G.; Gong, M. Small Hepatitis Delta Antigen Selectively Binds to Target mRNA in Hepatic Cells: A Potential Mechanism by Which Hepatitis D Virus Downregulates Glutathione S-Transferase P1 and Induces Liver Injury and Hepatocarcinogenesis. Biochem. Cell Biol. 2019, 97, 130–139. [Google Scholar] [CrossRef]
- Liao, F.T.; Lee, Y.J.; Ko, J.L.; Tsai, C.C.; Tseng, C.J.; Sheu, G.T. Hepatitis Delta Virus Epigenetically Enhances Clusterin Expression via Histone Acetylation in Human Hepatocellular Carcinoma Cells. J. Gen. Virol. 2009, 90, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Karna, R. A Review of the Diagnosis and Management of Hepatitis E. Curr. Treat. Options Infect. Dis. 2020, 12, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, R.; Morach, M.; Hübschke, E.; Bachofen, C.; Stephan, R.; Nüesch-Inderbinen, M. Seroprevalence of Hepatitis E Virus in Dogs in Switzerland. Zoonoses Public Health 2021, 68, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.W.; Dalton, H.R. Hepatitis E: An Underestimated Emerging Threat. Ther. Adv. Infect. Dis. 2019, 6, 2049936119837162. [Google Scholar] [CrossRef]
- Mushahwar, I.K. Hepatitis E Virus: Molecular Virology, Clinical Features, Diagnosis, Transmission, Epidemiology, and Prevention. J. Med. Virol. 2008, 80, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Pischke, S.; Suneetha, P.V.; Baechlein, C.; Barg-Hock, H.; Heim, A.; Kamar, N.; Schlue, J.; Strassburg, C.P.; Lehner, F.; Raupach, R.; et al. Hepatitis E Virus Infection as a Cause of Graft Hepatitis in Liver Transplant Recipients. Liver Transpl. 2010, 16, 74–82. [Google Scholar] [CrossRef]
- Blasco-Perrin, H.; Madden, R.; Stanley, A.; Crossan, C.; Hunter, J.; Vine, L.; Lane, K.; Devooght-Johnson, N.; McLaughlin, C.; Petrik, J.; et al. Hepatitis E Virus in Patients with Decompensated Chronic Liver Disease: A Prospective UK/French Study. Aliment Pharmacol. Ther. 2015, 42, 574–581. [Google Scholar] [CrossRef]
- Borentain, P.; Colson, P.; Bolon, E.; Gauchez, P.; Coso, D.; Gérolami, R. Hepatocellular Carcinoma Complicating Hepatitis E Virus-Related Cirrhosis. Hepatology 2018, 67, 446–448. [Google Scholar] [CrossRef]
- Tseng, T.C.; Liu, C.J.; Chang, C.T.; Su, T.H.; Yang, W.T.; Tsai, C.H.; Chen, C.L.; Yang, H.C.; Liu, C.H.; Chen, P.J.; et al. HEV Superinfection Accelerates Disease Progression in Patients with Chronic HBV Infection and Increases Mortality in Those with Cirrhosis. J. Hepatol. 2020, 72, 1105–1111. [Google Scholar] [CrossRef]
- Atsama, M.A.; Atangana, P.J.A.; Noah, D.N.; Moundipa, P.F.; Pineau, P.; Njouom, R. Hepatitis E Virus Infection as a Promoting Factor for Hepatocellular Carcinoma in Cameroon: Preliminary Observations. Int. J. Infect. Dis. 2017, 64, 4–8. [Google Scholar] [CrossRef]
- Owusu, M.; Bonney, J.K.; Annan, A.A.; Mawuli, G.; Okyere, K.; Mutocheluh, M.; Aryeequaye, J.; Adjei, N.K.; Afihene, M.; Spangenberg, K.; et al. Aetiology of Viral Hepatitis among Jaundiced Patients Presenting to a Tertiary Hospital in Ghana. PLoS ONE 2018, 13, e0203699. [Google Scholar] [CrossRef]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular Carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Kanda, T.; Yokosuka, O.; Omata, M. Hepatitis C Virus and Hepatocellular Carcinoma. Biology 2013, 2, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Pavio, N.; Aggarwal, R.; Labrique, A.; Wedemeyer, H.; Dalton, H.R. Hepatitis E Virus Infection. Nat. Rev. Dis. Prim. 2017, 3, 17086. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Bendall, R.; Legrand-Abravanel, F.; Xia, N.S.; Ijaz, S.; Izopet, J.; Dalton, H.R. Hepatitis E. Lancet 2012, 379, 2477–2488. [Google Scholar] [CrossRef]
- Mallet, V.; Nicand, E.; Sultanik, P.; Chakvetadze, C.; Tessé, S.; Thervet, E.; Mouthon, L.; Sogni, P.; Pol, S. Brief Communication: Case Reports of Ribavirin Treatment for Chronic Hepatitis E. Ann. Intern. Med. 2010, 153, 85–89. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef]
- Trinchet, J.C.; Bourcier, V.; Chaffaut, C.; Ait Ahmed, M.; Allam, S.; Marcellin, P.; Guyader, D.; Pol, S.; Larrey, D.; De Lédinghen, V.; et al. Complications and Competing Risks of Death in Compensated Viral Cirrhosis (ANRS CO12 CirVir Prospective Cohort). Hepatology 2015, 62, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef]
- Terrault, N.A.; Lok, A.S.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference | Factor | Location of Mutation |
|---|---|---|
| [42] | HBV serum level | |
| [45,50] | Genotype C | |
| [51] | Combined HbsAg/HbeAg positivity | |
| [52,54] | T1762/A1764 mutation | Basal core promoter |
| [46,55] | C1653T/T1753V mutation | Hbx |
| [61,62] | K130M/V131I/V131I mutation | Hbx |
| [66] | PreS deletion | PreS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datfar, T.; Doulberis, M.; Papaefthymiou, A.; Hines, I.N.; Manzini, G. Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens 2021, 10, 1366. https://doi.org/10.3390/pathogens10111366
Datfar T, Doulberis M, Papaefthymiou A, Hines IN, Manzini G. Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens. 2021; 10(11):1366. https://doi.org/10.3390/pathogens10111366
Chicago/Turabian StyleDatfar, Toofan, Michael Doulberis, Apostolis Papaefthymiou, Ian N. Hines, and Giulia Manzini. 2021. "Viral Hepatitis and Hepatocellular Carcinoma: State of the Art" Pathogens 10, no. 11: 1366. https://doi.org/10.3390/pathogens10111366
APA StyleDatfar, T., Doulberis, M., Papaefthymiou, A., Hines, I. N., & Manzini, G. (2021). Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens, 10(11), 1366. https://doi.org/10.3390/pathogens10111366