Crystal Structures of Al2Cu Revisited: Understanding Existing Phases and Exploring Other Potential Phases


Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
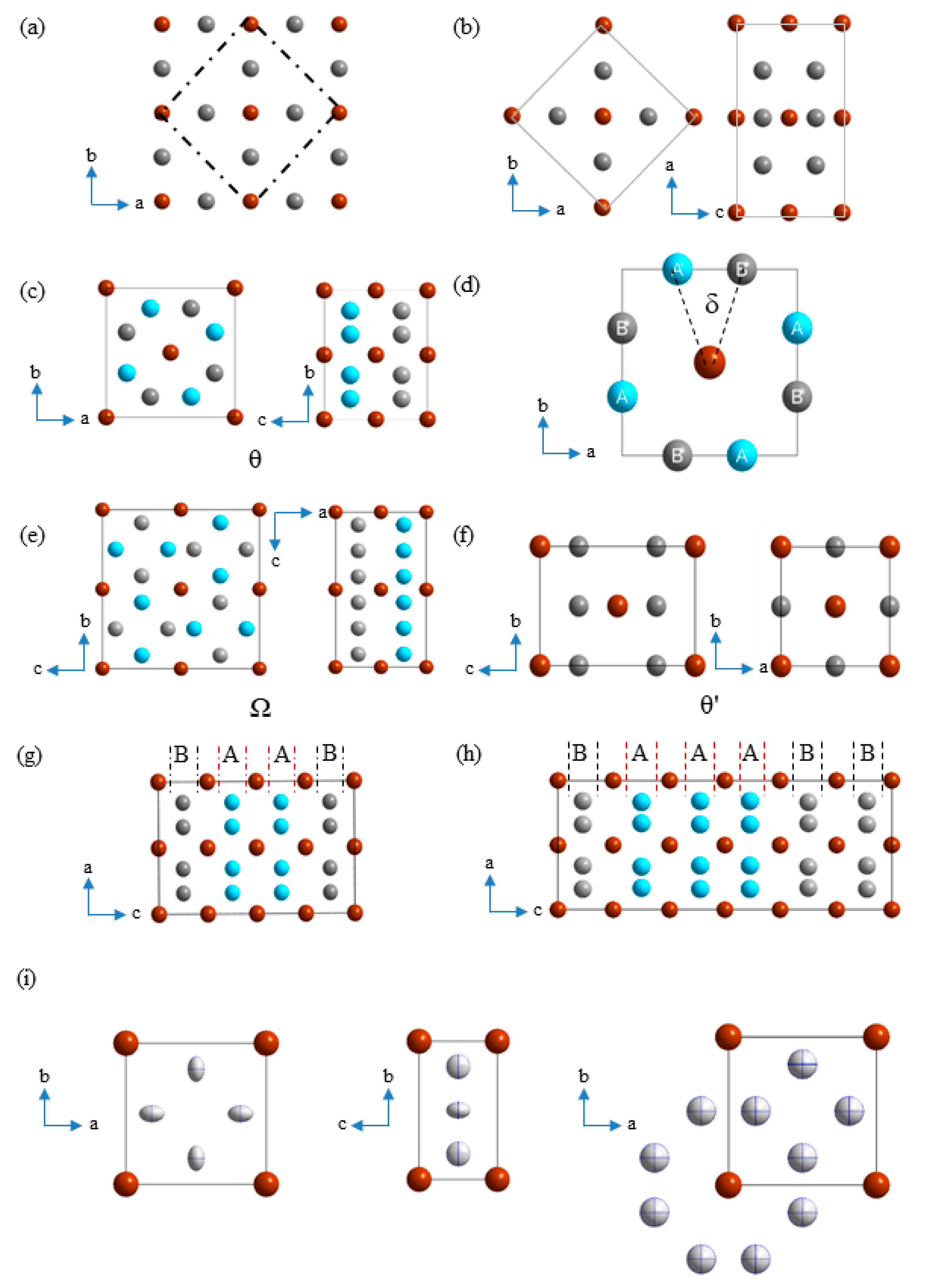
3.1. Structural Relations of the Existing Phases
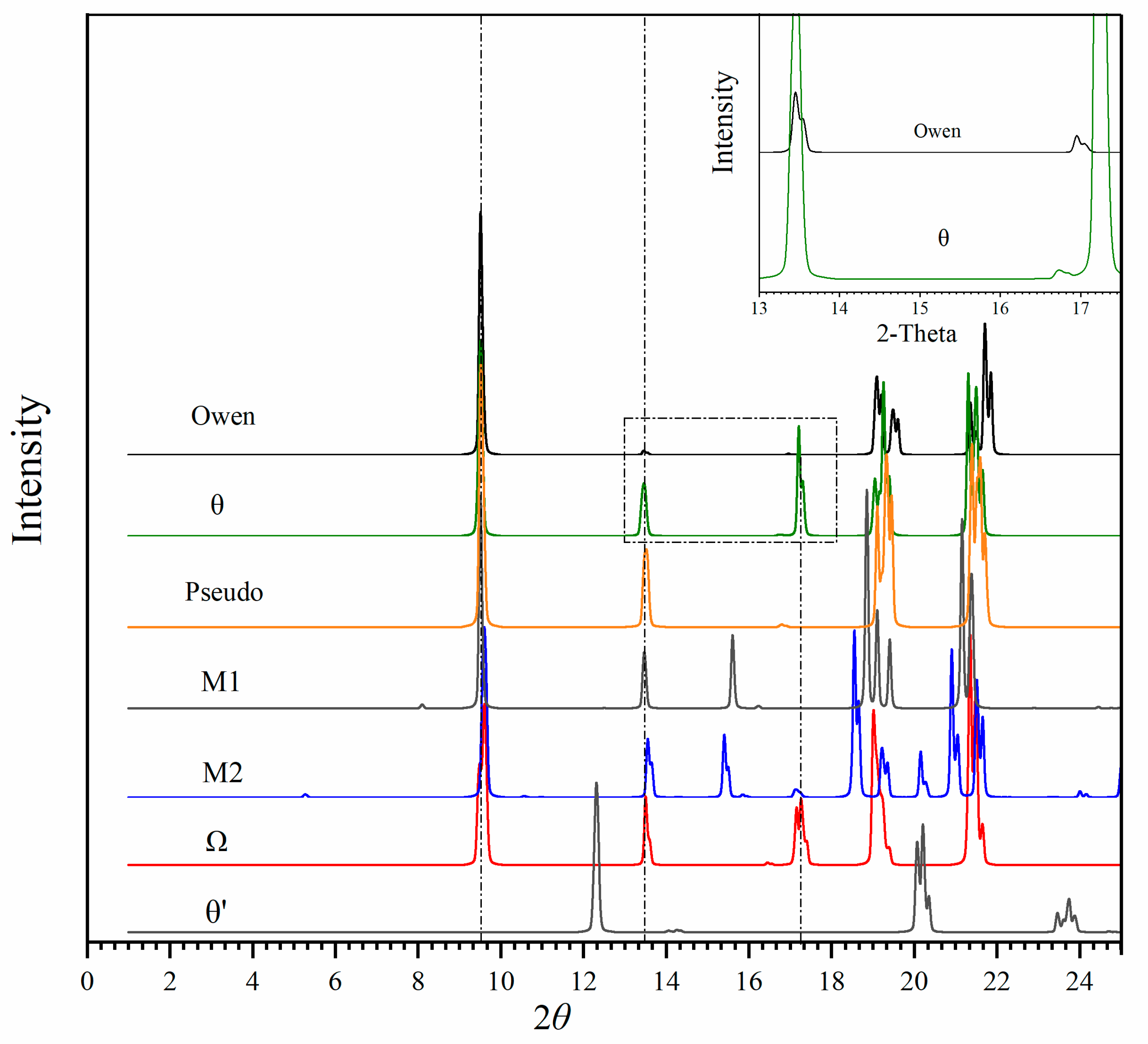
3.2. Potential Phases and Diffraction Patterns
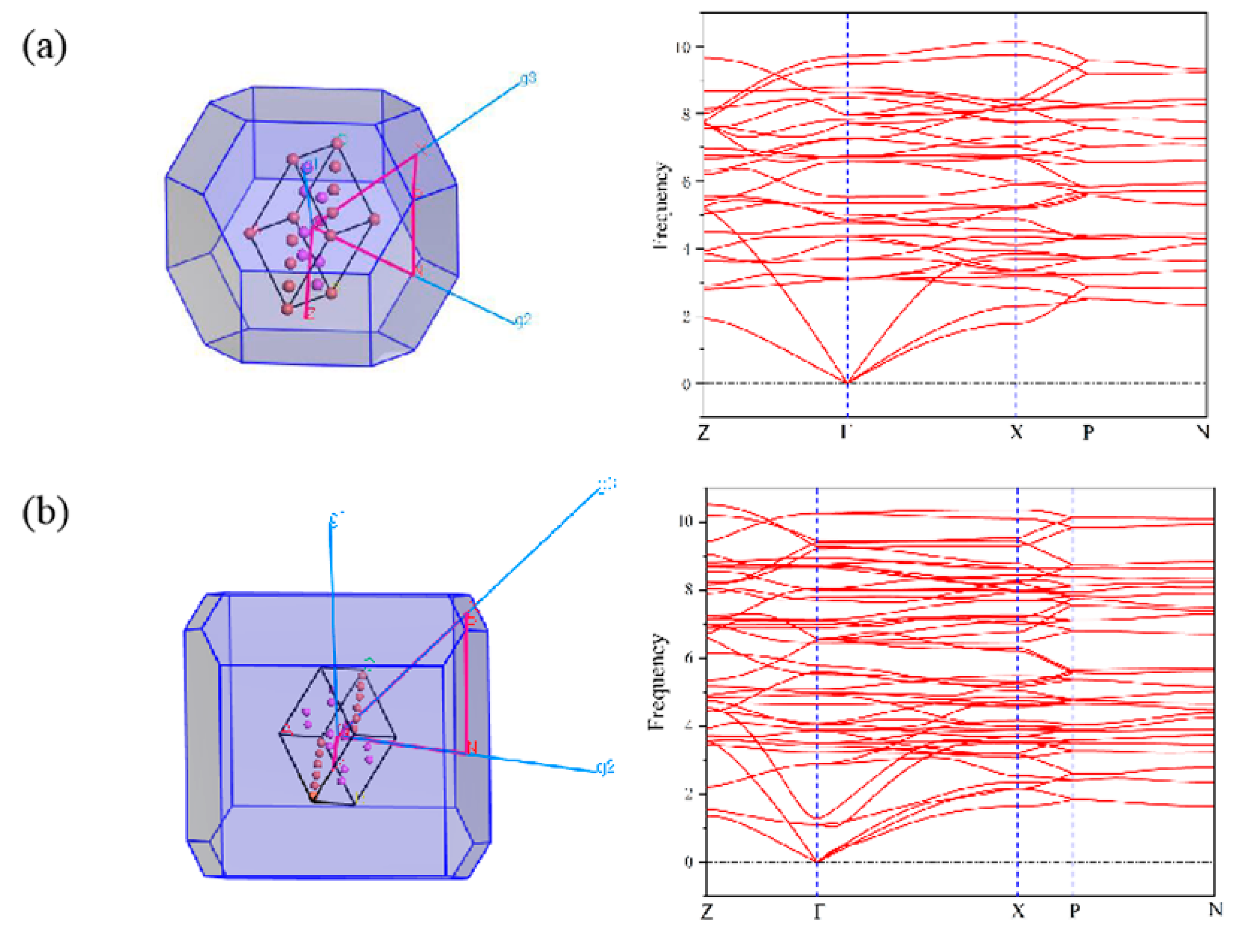
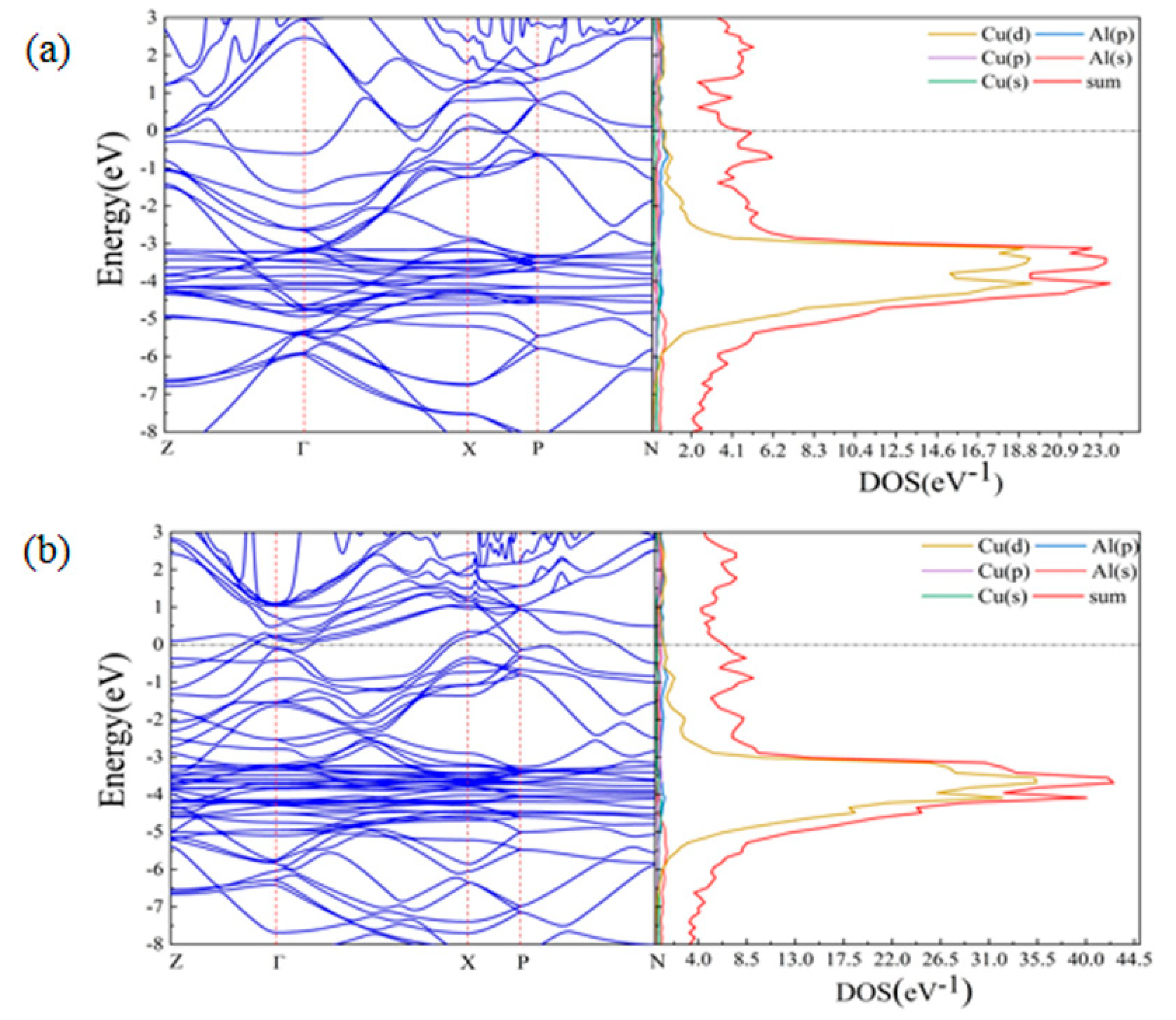
3.3. First-Principles Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raza, M.R.; Ahmad, F.; Ikram, N.; Ahmad, R.; Salam, A. Development and strengthening of 2219 aluminium alloy by mechanical working and heat treatment. J. Appl. Sci. 2011, 11, 1857–1861. [Google Scholar] [CrossRef]
- Owen, E.A.; Preston, G.D. X-ray analysis of solid solutions. Proc. Phys. Soc. Lond. 1923, 36, 14–30. [Google Scholar] [CrossRef]
- Jette, E.R.; Phragmen, G.; Westgren, A.F. X-Ray Studies on the Copper-Aluminium Alloys. J. Inst. Met. Lond. Bd. 1924, 31, 193–215. [Google Scholar]
- Friauf, J.B. The crystal structures of two intermetallic compounds. J. Am. Chem. Soc. 1927, 49, 3107–3114. [Google Scholar] [CrossRef]
- Meetsma, A.; De Boer, J.L.; Van Smaalen, S. Refinement of the Crystal Structure of Tetragonal Al2Cu. J. Solid State Chem. 1989, 83, 370–372. [Google Scholar] [CrossRef]
- Grin, Y.; Wagner, F.R.; Armbrüster, M.; Kohout, M.; Leithe-Jasper, A.; Schwarz, U.; Wedig, U.; Georgvon Schnering, H. CuAl2 revisited: Composition, crystal structure, chemical bonding, compressibility and raman spectroscopy. Solid State Chem. 2006, 179, 1707–1719. [Google Scholar] [CrossRef]
- Chen, H.C.; Yang, L.J.; Long, J.P. First-principles investigation of the elastic, Vickers hardness and thermodynamic properties of Al–Cu intermetallic compounds. Superlattice Microst. 2015, 79, 156–165. [Google Scholar] [CrossRef]
- Gayle, F.W.; Goodway, M. Precipitation hardening in the first aerospace aluminum alloy: The wright flyer crankcase. Science 1994, 266, 1015–1017. [Google Scholar] [CrossRef]
- Silcock, J.M.; Heal, T.J.; Hardy, H.K. Structural ageing characteristics of binary aluminium-copper alloys. J. Inst. Met. 1953, 82, 239–248. [Google Scholar]
- Knowles, K.; Stobbs, W. The structure of {111} age-hardening precipitates in Al–Cu-Mg-Ag alloys. Acta Cryst. 1988, B44, 207–227. [Google Scholar] [CrossRef]
- Auld, J.H. Structure of a metastable precipitate in an Al–Cu-Mg-Ag alloy. Acta Cryst. 1972, A28, S98. [Google Scholar] [CrossRef]
- Kerry, S.; Scott, V.D. Structure and orientation relationship of precipitates formed in AI-Cu-Mg-Ag alloys. Met. Sci. 1984, 18, 289–294. [Google Scholar] [CrossRef]
- Kang, S.J.; Kim, Y.W.; Kim, M.; Zuo, J.M. Determination of interfacial atomic structure, misfits and energetics of Ω phase in Al–Cu–Mg–Ag alloy. Acta Mater. 2014, 81, 501–511. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, L.; Li, B.; Song, Q.; Wu, P. Structural, Elastic, and Electronic Properties of Al–Cu intermetallics from First-Principles Calculations. J. Electron. Mater. 2009, 38, 356–364. [Google Scholar] [CrossRef]
- Sun, D.; Wang, Y.; Zhang, X.; Zhang, M.; Niu, Y. First-principles calculation on the thermodynamic and elastic properties of precipitations in Al–Cu alloys. Superlattices Microst. 2016, 100, 112–119. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11181. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G. Density functional theory. Ann. Rev. Phys. Cherm. 1983, 34, 631–656. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106(1)–134106(9). [Google Scholar] [CrossRef]
- Sahu, B.R. Electronic structure and bonding of ultralight LiMg. Mater. Sci. Eng. B 1997, 49, 74–78. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Hou, H.; Han, P. First-principles investigation of the structural, mechanical and thermodynamic properties of Al2Cu phase under various pressure and temperature conditions. Solid State Commun. 2017, 268, 44–50. [Google Scholar] [CrossRef]
- Wolverton, C.; Ozoliņš, V. Entropically Favored Ordering: The Metallurgy of Al2Cu Revisited. Phys. Rev. Lett. 2001, 86, 5518(1)–5518(4). [Google Scholar] [CrossRef] [PubMed]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B. 2014, 90, 224104(1)–224104(4). [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Space Group | Lattice Parameters | Wyckoff Positions | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Symbol | Number | a | b | c | Atom Site | x | y | z | ||
| Owen a | P4/mmm | 123 | 4.28 | 4.28 | 2.405 | Al1 | 2e | 1/2 | 0 | 1/2 |
| Cu1 | 1a | 0 | 0 | 0 | ||||||
| Ω b | Fmmm | 69 | 4.96 | 8.59 | 8.48 | Al1 | 8h | 3/4 | 1/12 | 3/4 |
| Al2 | 8i | 3/4 | 3/4 | 11/12 | ||||||
| Cu1 | 8f | 0 | 0 | 0 | ||||||
| θ c | I4/mcm | 140 | 6.067 | 6.067 | 4.877 | Al1 | 8h | 0.1581 | 0.6581 | 0 |
| Cu1 | 4a | 0 | 0 | 1/4 | ||||||
| θ’ d | I4/mmm | 139 | 4.04 | 4.04 | 5.80 | Al1 | 4d | 1/2 | 0 | 1/4 |
| Cu1 | 2a | 0 | 0 | 0 | ||||||
| M1 e | I4/mcm | 140 | 6.047 | 6.047 | 10.052 | Al1 | 16l | 0.8405 | 0.3405 | 0.8855 |
| Cu1 | 4c | 0 | 0 | 1/4 | ||||||
| Cu2 | 4a | 0 | 0 | 0 | ||||||
| M2 e | I4/mcm | 140 | 6.008 | 6.008 | 15.445 | Al1 | 16l | 0.8406 | 0.3406 | 0.5744 |
| Al2 | 8h | 0.8320 | 0.3320 | 3/4 | ||||||
| Cu1 | 8f | 0 | 0 | 0.1654 | ||||||
| Cu2 | 4a | 0 | 0 | 0 | ||||||
| Pseudo e | P4/mmm | 123 | 4.275 | 4.275 | 2.431 | Al1 | 4l | 0.8159 | 1/2 | 1/2 |
| Cu1 | 1d | 0 | 0 | 0 | ||||||
| Phase | Lattice Constants (Å) | C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 | ΔH(eV/atom) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | |||||||||||
| Owen | 4.11 | 4.11 | 2.89 | 195.8 | 152.7 | 40.1 | 39.9 | 26.7 | 64.5 | −0.025 | |||
| θ a,b | 6.05 | 6.05 | 4.87 | 156.3 | - | 170.2 | 31.9 | - | 40.9 | 83.2 | 65.3 | −0.153 | |
| θ’ c,b | 4.04 | 4.04 | 5.80 | 158.9 | - | 160.3 | 48.0 | - | 40.5 | 56.8 | 60.9 | −0.167 | |
| Ω d,b | 4.96 | 8.56 | 8.48 | 161.7 | 159.2 | 164.2 | 42.5 | 41.9 | 49.9 | 65.7 | 69.6 | 80.8 | −0.154 |
| M1 | 6.04 | 6.04 | 10.0 | 81.7 | - | 176.0 | 31.6 | - | 42.9 | 27.3 | 53.0 | - | −0.124 |
| M2 | 6.00 | 6.00 | 15.44 | 99.7 | - | 147.9 | 18.6 | - | 36.3 | 32.9 | 43.4 | - | −0.108 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Fan, C. Crystal Structures of Al2Cu Revisited: Understanding Existing Phases and Exploring Other Potential Phases. Metals 2019, 9, 1037. https://doi.org/10.3390/met9101037
Wang S, Fan C. Crystal Structures of Al2Cu Revisited: Understanding Existing Phases and Exploring Other Potential Phases. Metals. 2019; 9(10):1037. https://doi.org/10.3390/met9101037
Chicago/Turabian StyleWang, Sai, and Changzeng Fan. 2019. "Crystal Structures of Al2Cu Revisited: Understanding Existing Phases and Exploring Other Potential Phases" Metals 9, no. 10: 1037. https://doi.org/10.3390/met9101037
APA StyleWang, S., & Fan, C. (2019). Crystal Structures of Al2Cu Revisited: Understanding Existing Phases and Exploring Other Potential Phases. Metals, 9(10), 1037. https://doi.org/10.3390/met9101037