Stability, Electronic Structure, and Dehydrogenation Properties of Pristine and Doped 2D MgH2 by the First Principles Study

Abstract
1. Introduction
2. Computational Details
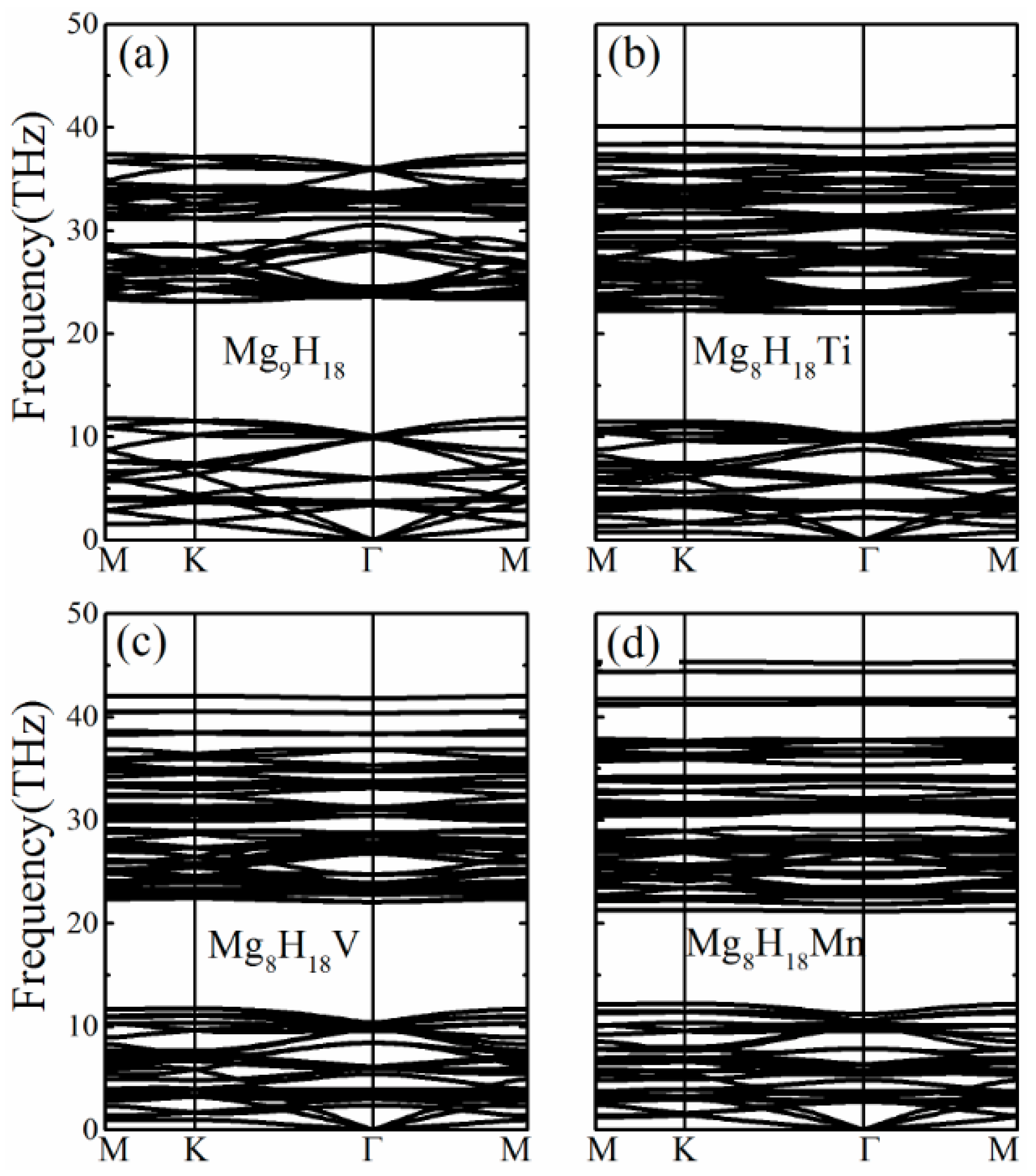
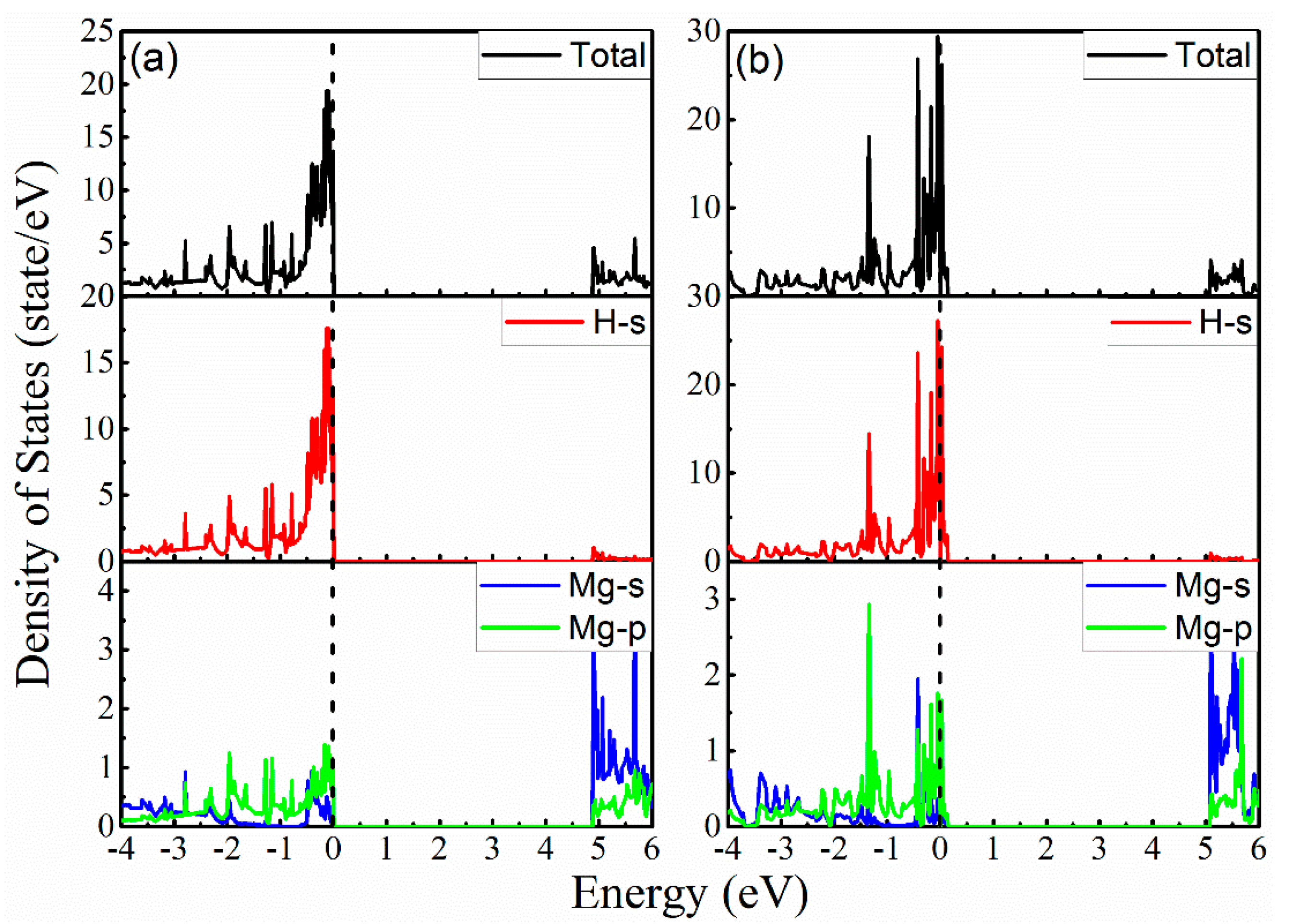
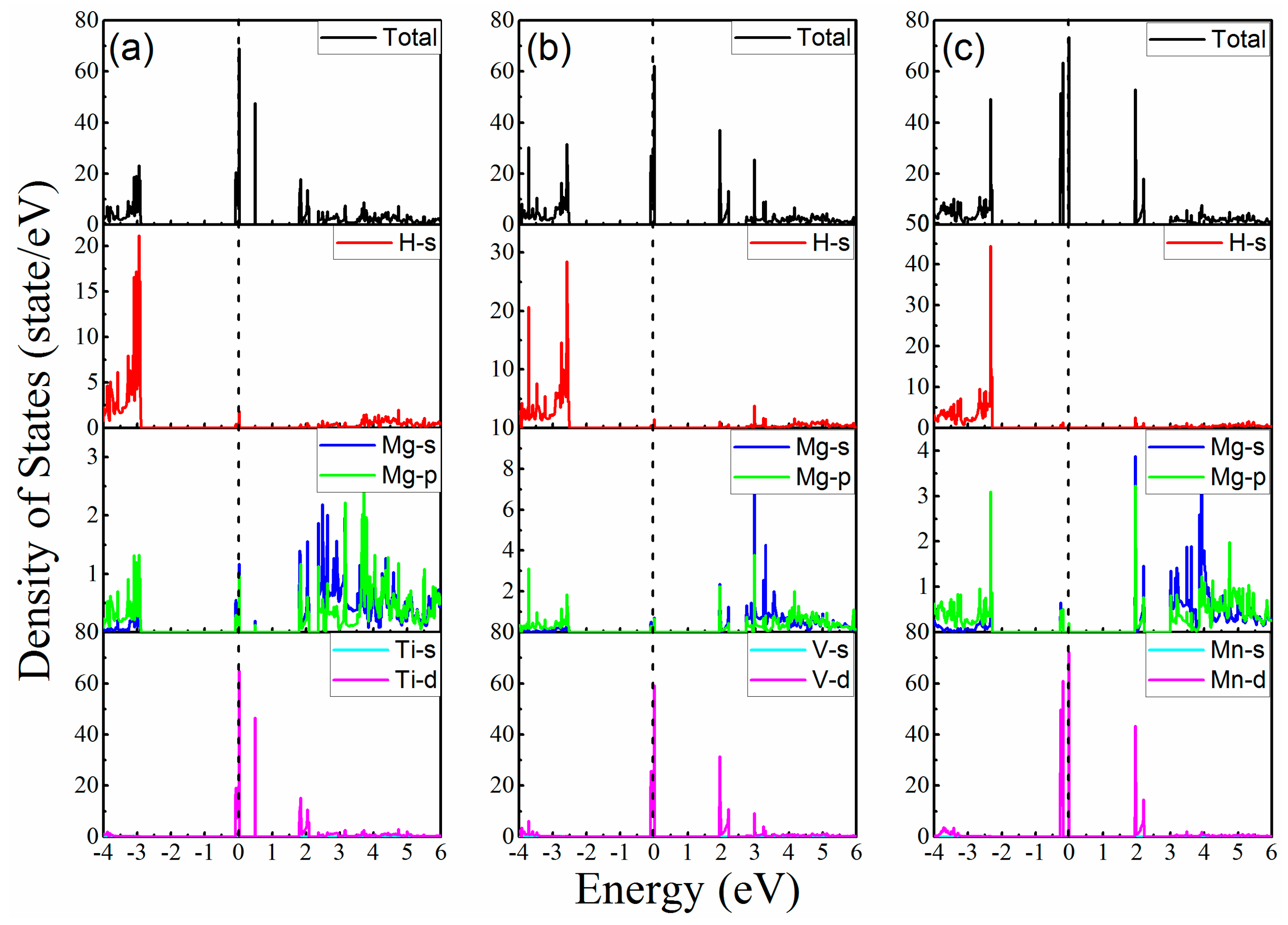
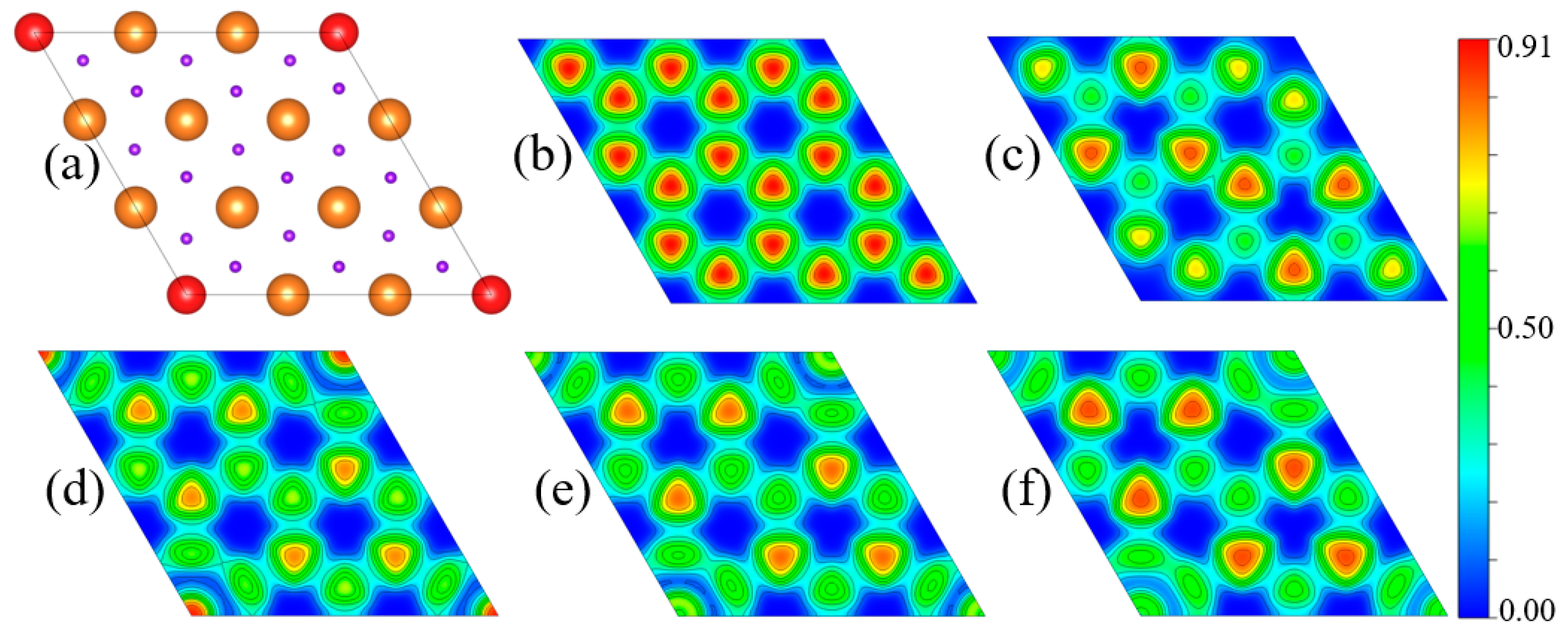
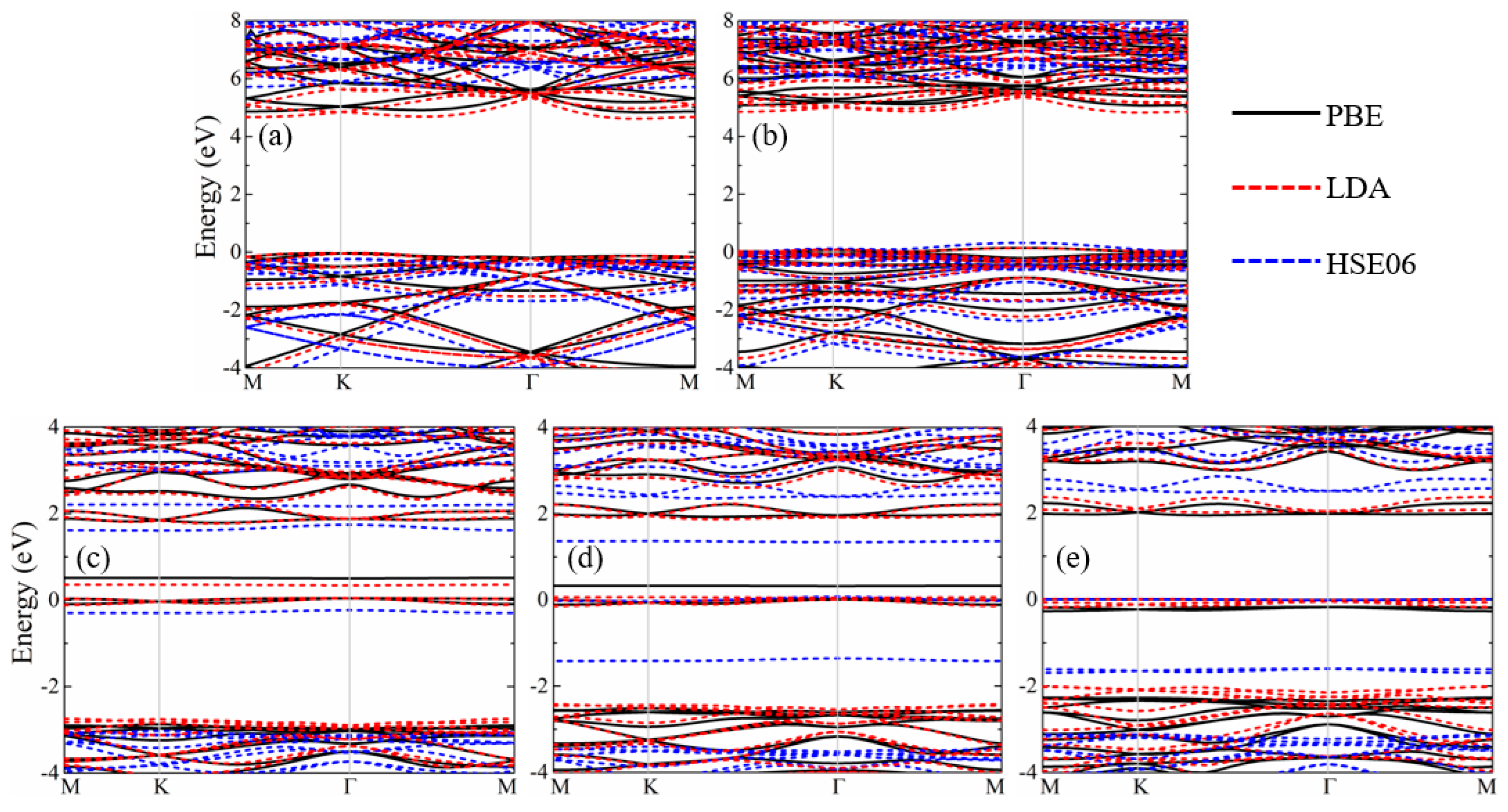
3. Results and Discussion
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydride | PBE | LDA | ||||||
|---|---|---|---|---|---|---|---|---|
| a (Å) | α (°) | β (°) | γ (°) | a (Å) | α (°) | β (°) | γ (°) | |
| Mg9H18 | 9.033 | 90.0 | 90.0 | 120.0 | 8.894 | 90.0 | 90.0 | 120.0 |
| Mg8H18 | 9.062 | 90.0 | 90.0 | 120.0 | 8.888 | 90.0 | 90.0 | 120.0 |
| Mg8H18Ti | 9.027 | 90.0 | 90.0 | 120.0 | 8.883 | 90.0 | 90.0 | 120.0 |
| Mg8H18V | 8.951 | 90.0 | 90.0 | 120.0 | 8.803 | 90.0 | 90.0 | 120.0 |
| Mg8H18Mn | 8.815 | 90.0 | 90.0 | 120.0 | 8.662 | 90.0 | 90.0 | 120.0 |

References
- Sakintuna, B.; Lamari-Darkrim, F.; Hirscher, M. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen Energy 2007, 32, 1121–1140. [Google Scholar] [CrossRef]
- Mohammed, Z.; Ahmed, R.; Mohammed Benali, K.; Bakhtiar ul, H.; Ahmad Radzi Mat, I.; Souraya, G.-S. First principle investigations of the physical properties of hydrogen-rich MgH2. Phys. Scr. 2013, 88, 065704. [Google Scholar] [CrossRef]
- Shang, C.X.; Bououdina, M.; Song, Y.; Guo, Z.X. Mechanical alloying and electronic simulations of (MgH2 + M) systems (M = Al, Ti, Fe, Ni, Cu and Nb) for hydrogen storage. Int. J. Hydrogen Energy 2004, 29, 73–80. [Google Scholar] [CrossRef]
- Ul Haq, B.; Kanoun, M.B.; Ahmed, R.; Bououdina, M.; Goumri-Said, S. Hybrid functional calculations of potential hydrogen storage material: Complex dimagnesium iron hydride. Int. J. Hydrogen Energy 2014, 39, 9709–9717. [Google Scholar] [CrossRef]
- Kumar, D.; Singh, A.; Prasad Tiwari, G.; Kojima, Y.; Kain, V. Thermodynamics and kinetics of nano-engineered Mg-MgH2 system for reversible hydrogen storage application. Thermochim. Acta 2017, 652, 103–108. [Google Scholar] [CrossRef]
- Trivedi, D.R.; Bandyopadhyay, D. Study of adsorption and dissociation process of H2 molecule on MgnRh clusters: A density functional investigation. Int. J. Hydrogen Energy 2016, 41, 20113–20121. [Google Scholar] [CrossRef]
- Song, Y.; Guo, Z.X.; Yang, R. Influence of selected alloying elements on the stability of magnesium dihydride for hydrogen storage applications: A first-principles investigation. Phys. Rev. B 2004, 69, 094205. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Kjekshus, A.; Fjellvåg, H. Pressure-Induced Structural Transitions in MgH2. Phys. Rev. Lett. 2002, 89, 175506. [Google Scholar] [CrossRef] [PubMed]
- Kurko, S.; Matović, L.; Novaković, N.; Matović, B.; Jovanović, Z.; Mamula, B.P.; Grbović Novaković, J. Changes of hydrogen storage properties of MgH2 induced by boron ion irradiation. Int. J. Hydrogen Energy 2011, 36, 1184–1189. [Google Scholar] [CrossRef]
- Song, M.Y.; Kwon, S.N.; Park, H.R.; Hong, S.-H. Improvement in the hydrogen storage properties of Mg by mechanical grinding with Ni, Fe and V under H2 atmosphere. Int. J. Hydrogen Energy 2011, 36, 13587–13594. [Google Scholar] [CrossRef]
- Noritake, T.; Aoki, M.; Towata, S.; Seno, Y.; Hirose, Y.; Nishibori, E.; Takata, M.; Sakata, M. Chemical bonding of hydrogen in MgH2. Appl. Phys. Lett. 2002, 81, 2008–2010. [Google Scholar] [CrossRef]
- Liang, G.; Huot, J.; Boily, S.; Van Neste, A.; Schulz, R. Catalytic effect of transition metals on hydrogen sorption in nanocrystalline ball milled MgH2 − Tm (Tm = Ti, V, Mn, Fe and Ni) systems. J. Alloys Compd. 1999, 292, 247–252. [Google Scholar] [CrossRef]
- Shang, C.X.; Bououdina, M.; Guo, Z.X. Structural stability of mechanically alloyed (Mg + 10Nb) and (MgH2 + 10Nb) powder mixtures. J. Alloys Compd. 2003, 349, 217–223. [Google Scholar] [CrossRef]
- Rivoirard, S.; de Rango, P.; Fruchart, D.; Charbonnier, J.; Vempaire, D. Catalytic effect of additives on the hydrogen absorption properties of nano-crystalline MgH2(X) composites. J. Alloys Compd. 2003, 356, 622–625. [Google Scholar] [CrossRef]
- Oelerich, W.; Klassen, T.; Bormann, R. Metal oxides as catalysts for improved hydrogen sorption in nanocrystalline Mg-based materials. J. Alloys Compd. 2001, 315, 237–242. [Google Scholar] [CrossRef]
- Aguey-Zinsou, K.F.; Ares Fernandez, J.R.; Klassen, T.; Bormann, R. Effect of Nb2O5 on MgH2 properties during mechanical milling. Int. J. Hydrogen Energy 2007, 32, 2400–2407. [Google Scholar] [CrossRef]
- Song, M.; Bobet, J.-L.; Darriet, B. Improvement in hydrogen sorption properties of Mg by reactive mechanical grinding with Cr2O3, Al2O3 and CeO2. J. Alloys Compd. 2002, 340, 256–262. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Erratum: Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2006, 118, 8207. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-Principles Calculations of the Ferroelastic Transition Between Rutile-Type and CaCl2-Type SiO2 at High Pressures. Phys. Rev. B Condens. Matter 2008, 78. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355–10368. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Gu, T.; Wang, Z.; Tada, T.; Watanabe, S. First-principles simulations on bulk Ta2O5 and Cu/Ta2O5/Pt heterojunction: Electronic structures and transport properties. J. Appl. Phys. 2009, 106, 262907. [Google Scholar] [CrossRef]
- Sun, R.; Wang, Z.; Saito, M.; Shibata, N.; Ikuhara, Y. Atomistic mechanisms of nonstoichiometry-induced twin boundary structural transformation in titanium dioxide. Nat. Commun. 2011, 6, 7120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Saito, M.; Mckenna, K.P.; Gu, L.; Tsukimoto, S.; Shluger, A.L.; Ikuhara, Y. Atom-resolved imaging of ordered defect superstructures at individual grain boundaries. Nature 2011, 479, 380–383. [Google Scholar] [CrossRef] [PubMed]
- McKenna, K.P.; Hofer, F.; Gilks, D.; Lazarov, V.K.; Chen, C.; Wang, Z.; Ikuhara, Y. Atomic-scale structure and properties of highly stable antiphase boundary defects in Fe3O4. Nat. Commun. 2014, 5, 5740. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Saito, M.; Mckenna, K.P.; Fukami, S.; Sato, H.; Ikeda, S.; Ohno, H.; Ikuhara, Y. Atomic-Scale Structure and Local Chemistry of CoFeB-MgO Magnetic Tunnel Junctions. Nano Lett. 2016, 16, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Ziyu, H.; Xu, G.; Xiaohong, S. Structural, electronic and photocatalytic properties of atomic defective BiI3 monolayers. Chem. Phys. Lett. 2018, 691, 341–346. [Google Scholar] [CrossRef]
- García, G.N.; Abriata, J.P.; Sofo, J.O. Calculation of the electronic and structural properties of cubic Mg2NiH4. Phys. Rev. B 1999, 59, 11746–11754. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, J.; Xie, R.; Song, Y.; Bououdina, M. First principles study of dehydrogenation properties of alkali/alkali-earth metal doped Mg7TiH16. J. Alloys Compd. 2017, 728, 1016–1022. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Fruchart, D.; Wolfers, P. Electronic structure and stability of new FCC magnesium hydrides Mg7MH16 and Mg6MH16 (M = Ti, V, Nb): An ab initio study. Int. J. Hydrogen Energy 2010, 35, 2025–2032. [Google Scholar] [CrossRef]
- Dai, J.H.; Song, Y.; Yang, R. First Principles Study on Hydrogen Desorption from a Metal (=Al, Ti, Mn, Ni) Doped MgH2 (110) Surface. J. Phys. Chem. C 2010, 114, 11328–11334. [Google Scholar] [CrossRef]
- Kumar, M.; Kamal, R.; Thapa, R. Screening based approach and dehydrogenation kinetics for MgH2: Guide to find suitable dopant using first-principles approach OPEN. Sci. Rep. 2017, 7, 15550. [Google Scholar] [CrossRef] [PubMed]
- Van Mal, H.H.; Buschow, K.H.J.; Miedema, A.R. Hydrogen absorption in LaNi5 and related compounds: Experimental observations and their explanation. J. Less Common Met. 1974, 35, 65–76. [Google Scholar] [CrossRef]
- Lakhal, M.; Bhihi, M.; Benyoussef, A.; El Kenz, A.; Loulidi, M.; Naji, S. The hydrogen ab/desorption kinetic properties of doped magnesium hydride MgH2 systems by first principles calculations and kinetic Monte Carlo simulations. Int. J. Hydrogen Energy 2015, 40, 6137–6144. [Google Scholar] [CrossRef]
- Alapati, S.V.; Johnson, J.K.; Sholl, D.S. Identification of Destabilized Metal Hydrides for Hydrogen Storage Using First Principles Calculations. J. Phys. Chem. B 2006, 110, 8769–8776. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lam, P.K. Electronic and structural properties of MgH2. Phys. Rev. B 1988, 37, 8730–8737. [Google Scholar] [CrossRef]
- Westerwaal, R.J.; Broedersz, C.P.; Gremaud, R.; Slaman, M.; Borgschulte, A.; Lohstroh, W.; Tschersich, K.G.; Fleischhauer, H.P.; Dam, B.; Griessen, R. Study of the hydride forming process of in-situ grown MgH2 thin films by activated reactive evaporation. Thin Solid Films 2008, 516, 4351–4359. [Google Scholar] [CrossRef]





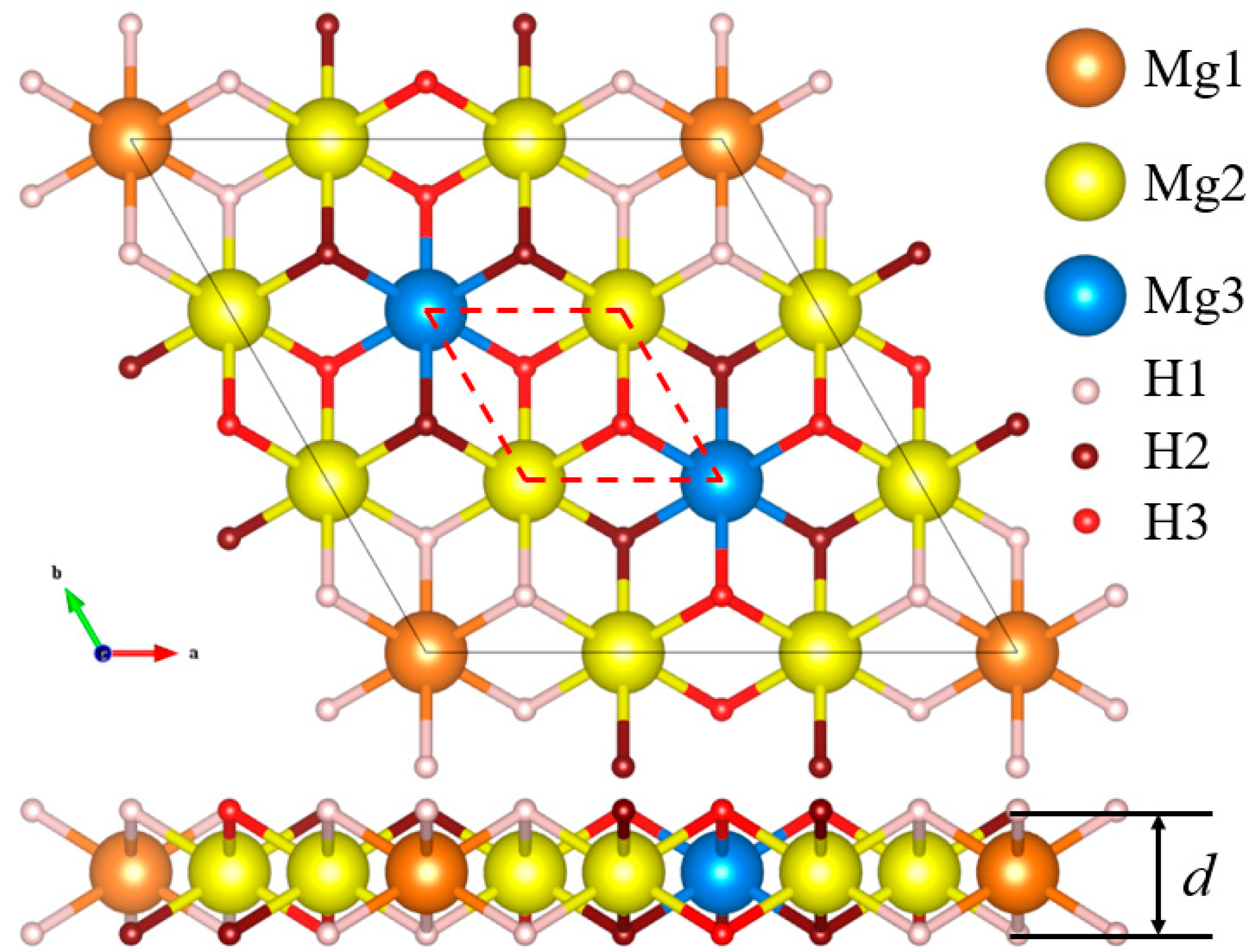
| Lattice Parameters | Atom | Wyckoff | Atomic Positions (Fractional) | ||
|---|---|---|---|---|---|
| Positions | x | y | z | ||
| 164(P-3m1) | Mg1 | 1b | 0 | 0 | 0.5 |
| a = b = 9.033 Å | Mg2 | 6h | 0 | 0.33333 | 0.5 |
| c = 15 Å | Mg3 | 2d | 0.33333 | 0.66667 | 0.5 |
| d = 1.86 Å | H1 | 6i | 0.11111 | 0.22222 | 0.43783 |
| α = β = 90° | H2 | 6i | 0.22222 | 0.44444 | 0.56217 |
| γ = 120° | H3 | 6i | 0.11111 | 0.55556 | 0.43783 |
| Hydride | ΔE (eV) | Parameter | Bond Length (Å) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mg1 | Mg2 | Mg3 | a (Å) | Sub-H1 | Mg2-H1 | Mg2-H2 | Mg2-H3 | Mg3-H2 | Mg3-H3 | |
| Mg9H18 | 0 | 0 | 0 | 9.033 | 1.972 | 1.972 | 1.972 | 1.972 | 1.972 | 1.972 |
| Mg8H18 | 2.968 | 2.968 | 2.968 | 9.062 | - | 1.894 | 2.043 | 1.976 | 1.947 | 1.992 |
| Mg8H18Ti | 1.113 | 1.114 | 1.114 | 9.027 | 1.915 | 1.997 | 1.964 | 1.982 | 1.946 | 1.990 |
| Mg8H18V | 1.818 | 1.818 | 1.819 | 8.951 | 1.822 | 1.999 | 1.945 | 1.992 | 1.939 | 1.983 |
| Mg8H18Mn | 1.279 | 1.279 | 1.279 | 8.815 | 1.691 | 2.028 | 1.911 | 2.008 | 1.937 | 1.965 |
| Hydride | ΔH | T | Bader Charge (e) | Ed | ||
|---|---|---|---|---|---|---|
| (kJ/mol·H2) | (K) | Mg | X | H | (eV) | |
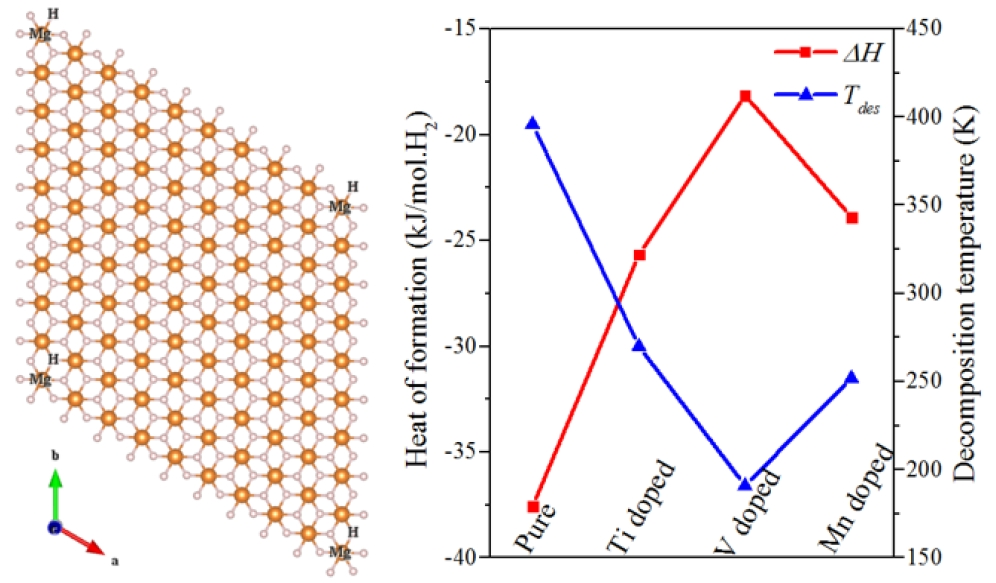
| Mg9H18 | −37.57 | 268~396 | +2.000 | - | −0.997 | 1.589 |
| Mg8H18 | 31.71 | - | +2.000 | - | −0.886 | −1.931 |
| Mg8H18Ti | −25.67 | 183~270 | +2.000 | +1.825 | −0.988 | 1.305 |
| Mg8H18V | −18.14 | 130~191 | +2.000 | +1.523 | −0.971 | 1.044 |
| Mg8H18Mn | −23.90 | 171~252 | +2.000 | +0.975 | −0.940 | 0.853 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, X.; Shao, X. Stability, Electronic Structure, and Dehydrogenation Properties of Pristine and Doped 2D MgH2 by the First Principles Study. Metals 2018, 8, 482. https://doi.org/10.3390/met8070482
Gong X, Shao X. Stability, Electronic Structure, and Dehydrogenation Properties of Pristine and Doped 2D MgH2 by the First Principles Study. Metals. 2018; 8(7):482. https://doi.org/10.3390/met8070482
Chicago/Turabian StyleGong, Xu, and Xiaohong Shao. 2018. "Stability, Electronic Structure, and Dehydrogenation Properties of Pristine and Doped 2D MgH2 by the First Principles Study" Metals 8, no. 7: 482. https://doi.org/10.3390/met8070482
APA StyleGong, X., & Shao, X. (2018). Stability, Electronic Structure, and Dehydrogenation Properties of Pristine and Doped 2D MgH2 by the First Principles Study. Metals, 8(7), 482. https://doi.org/10.3390/met8070482