Atomistic-Level Insights into MgO and Na2O Modifications of Molten Aluminosilicate Slag: A Molecular Dynamics Research on Structural Evolution and Properties



Abstract
1. Introduction
2. Simulation Approach
3. Results and Discussion
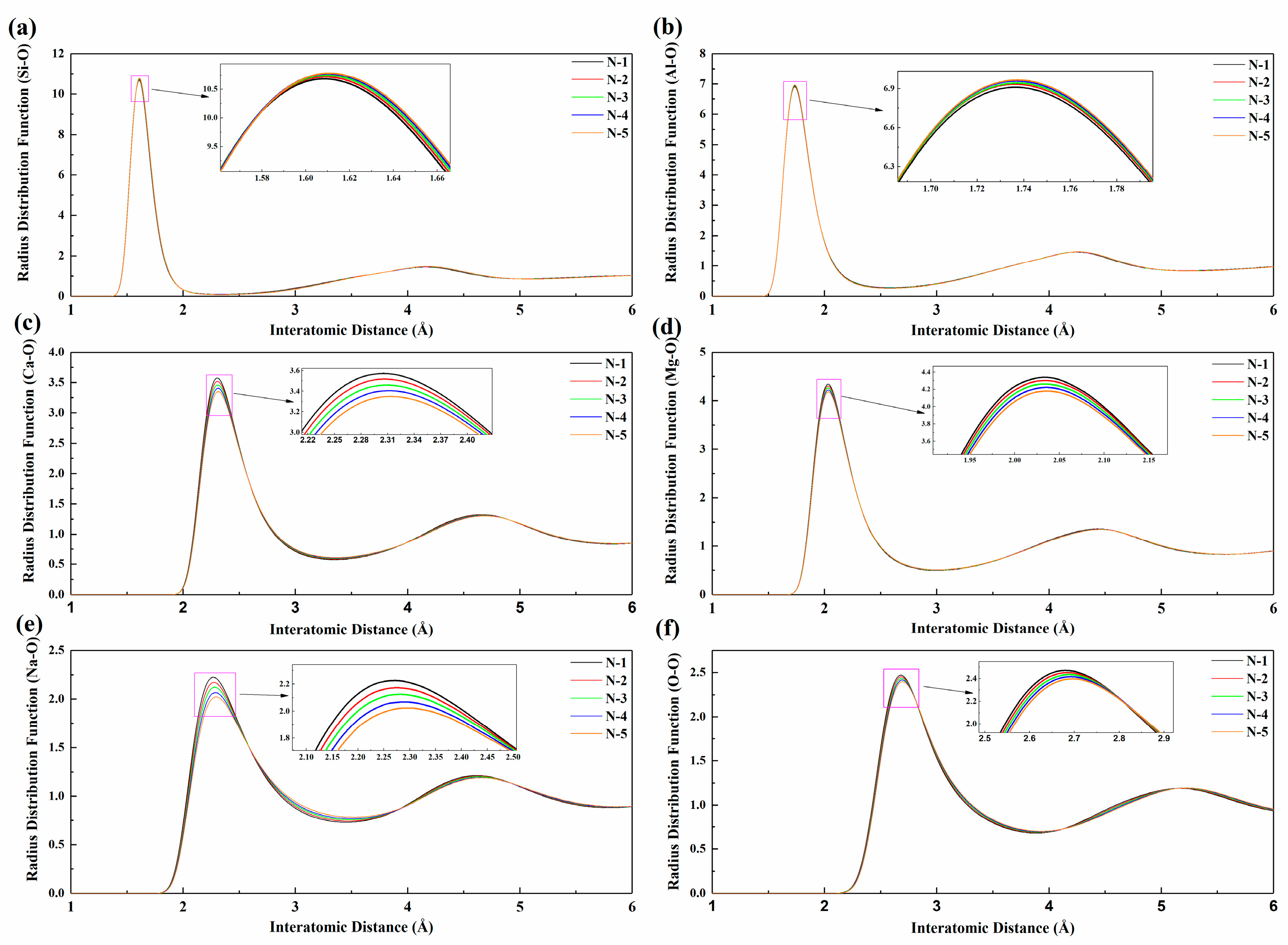
3.1. Local Structural Characteristics
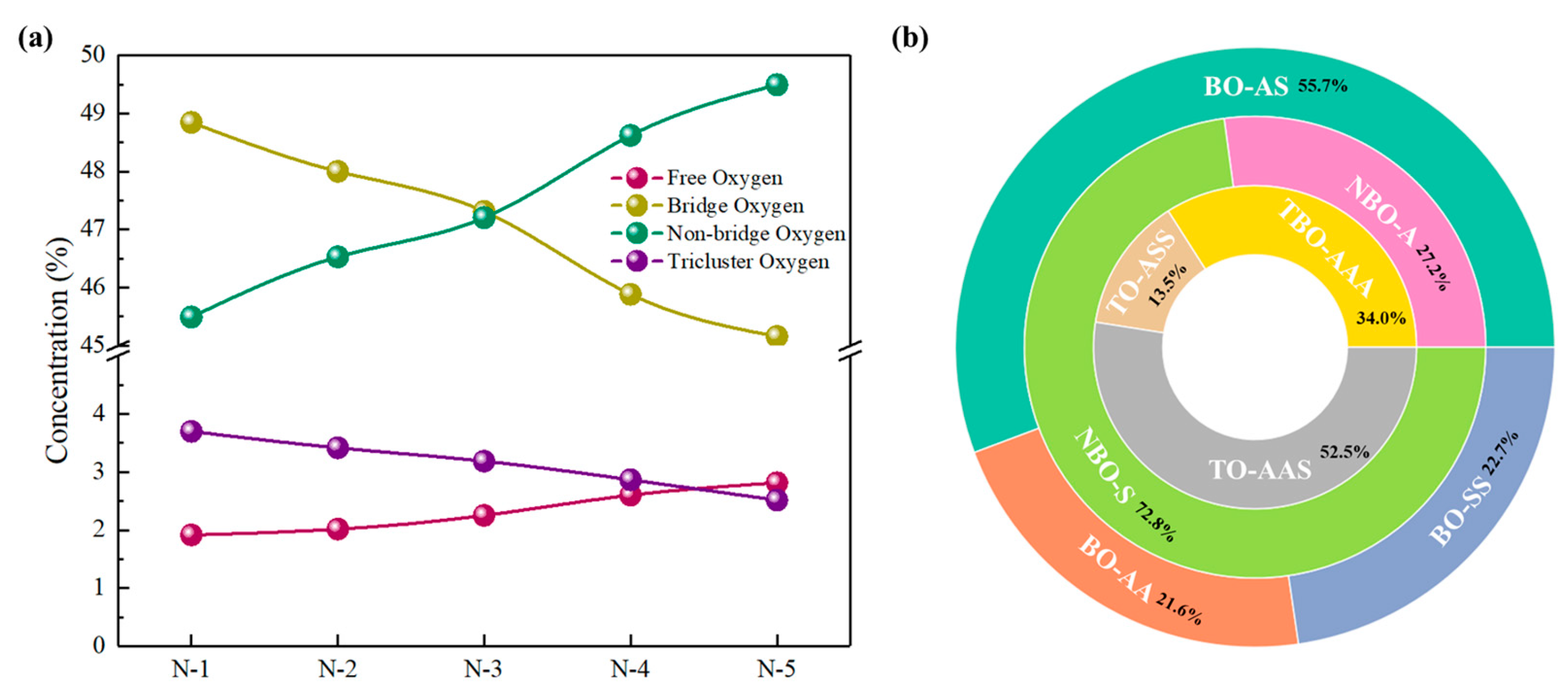
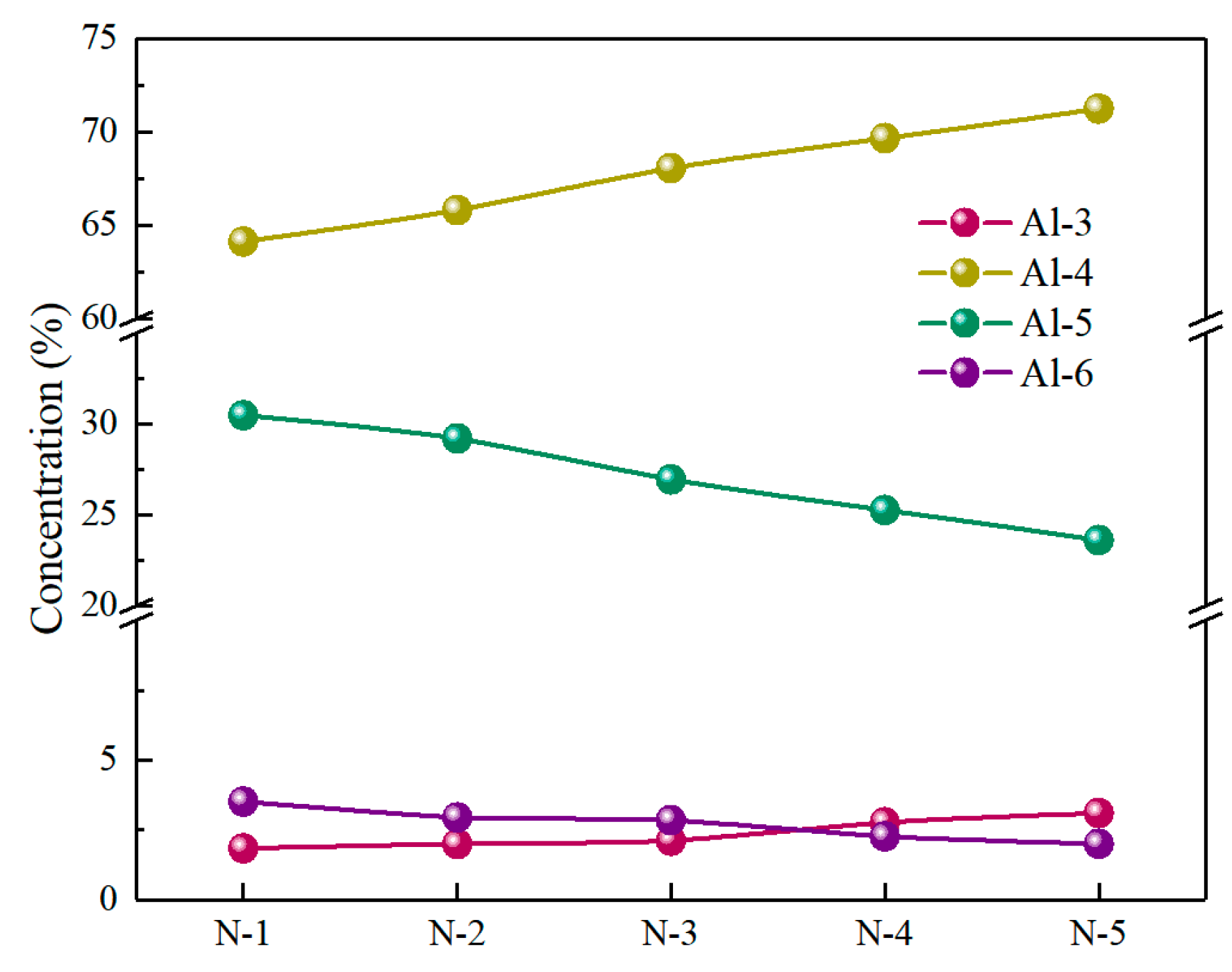
3.2. The Structural Unit Variation
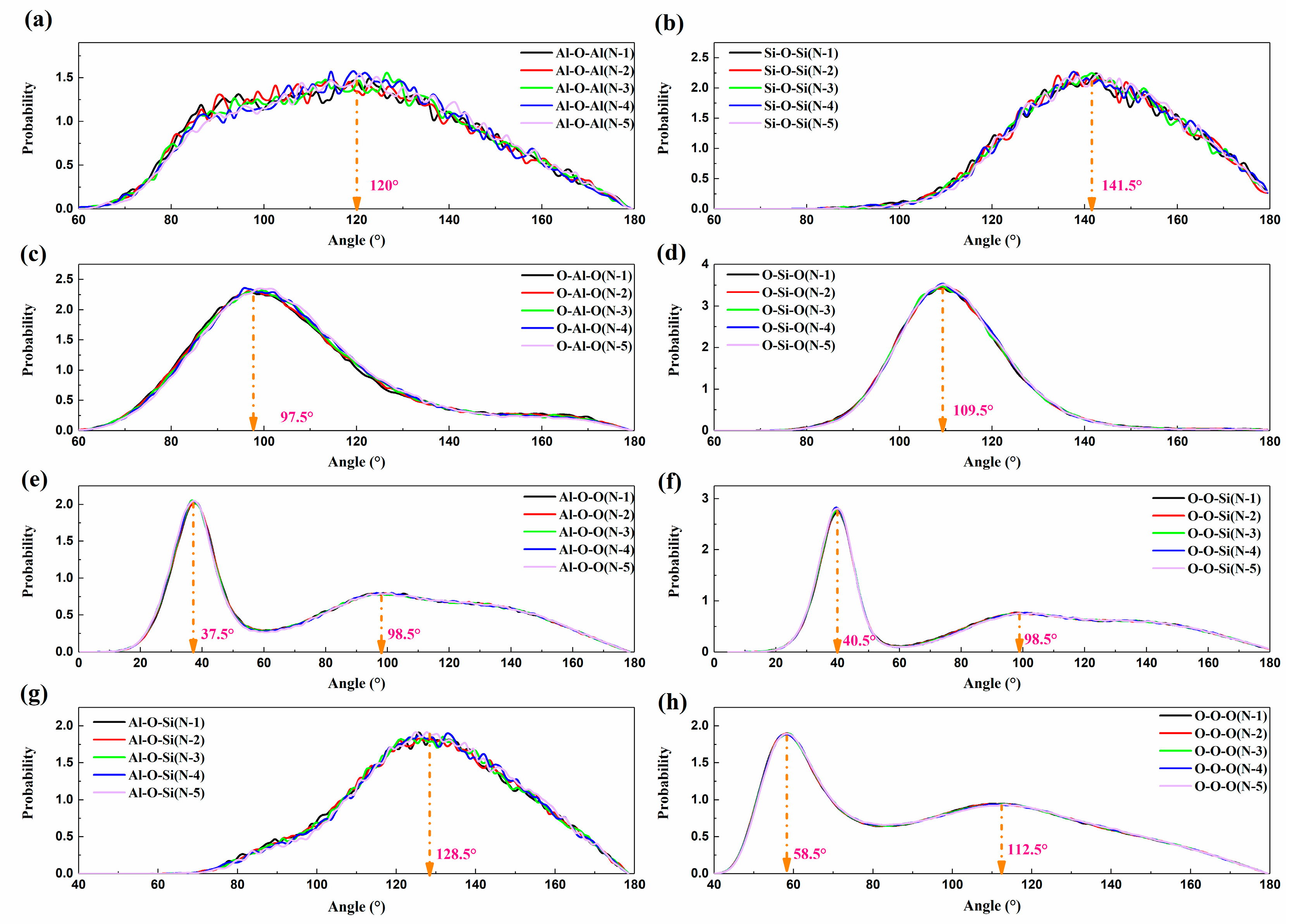
3.3. The Distribution of Bond Angle
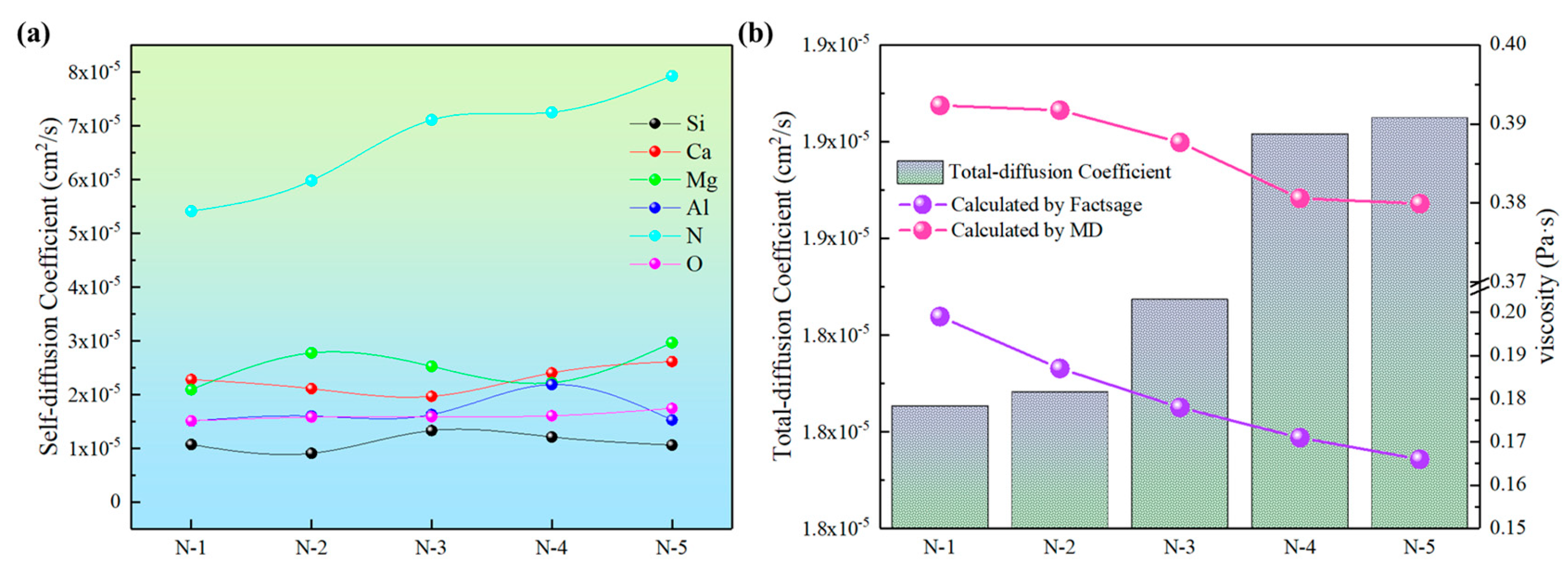
3.4. Transport Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yan, Z.-m.; Lv, X.-w.; Li, Z.-s. Physicochemical properties and structure of titania-containing metallurgical slags: A review. J. Iron Steel Res. Int. 2022, 29, 187–206. [Google Scholar] [CrossRef]
- Yasipourtehrani, S.; Strezov, V.; Bliznyukov, S.; Evans, T. Investigation of thermal properties of blast furnace slag to improve process energy efficiency. J. Clean. Prod. 2017, 149, 137–145. [Google Scholar] [CrossRef]
- Shi, C. Steel slag—Its production, processing, characteristics, and cementitious properties. J. Mater. Civ. Eng. 2004, 16, 230–236. [Google Scholar] [CrossRef]
- Mills, K.; Yuan, L.; Li, Z.; Zhang, G.; Chou, K. A review of the factors affecting the thermophysical properties of silicate slags. High Temp. Mater. Process. 2012, 31, 301–321. [Google Scholar] [CrossRef]
- Jiang, C.; Li, K.; Zhang, J.; Qin, Q.; Liu, Z.; Sun, M.; Wang, Z.; Liang, W. Effect of MgO/Al2O3 ratio on the structure and properties of blast furnace slags: A molecular dynamics simulation. J. Non-Cryst. Solids 2018, 502, 76–82. [Google Scholar] [CrossRef]
- Zhou, C.; Li, J.; Wang, S.; Zhao, J.; Ai, L.; Chen, Q.; Chen, Q.; Zhao, D. Development of molecular dynamics and research progress in the study of slag. Materials 2023, 16, 5373. [Google Scholar] [CrossRef]
- Liu, J.; Kong, W.; Yang, X.; Wang, Q.; He, Z.; Hou, X. Polymeric Structure Evolution Behavior Analysis of Aluminosilicate-Based Smelting Slag. Metals 2022, 12, 715. [Google Scholar] [CrossRef]
- Kim, T.S.; Park, J.H. Structure-viscosity relationship of low-silica calcium aluminosilicate melts. ISIJ Int. 2014, 54, 2031–2038. [Google Scholar] [CrossRef]
- Jiang, C.; Li, K.; Zhang, J.; Liu, Z.; Niu, L.; Liang, W.; Sun, M.; Ma, H.; Wang, Z. The effect of CaO and MgO on the structure and properties of coal ash in the blast furnace: A molecular dynamics simulation and thermodynamic calculation. Chem. Eng. Sci. 2019, 210, 115226. [Google Scholar] [CrossRef]
- Ji, X.; Wang, Z.; Wang, X.; Zhao, X.; Zhang, H.; Zhang, T. Microstructures and properties of alkali-activated slags with composite activator: Effects of Na2O equivalents. J. Clean. Prod. 2024, 450, 141754. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, H.; Xiong, Z.; Chen, S.; Li, K.; Zhang, J.; Liang, W.; Sun, M.; Wang, Z.; Wang, L. Molecular dynamics investigations on the effect of Na2O on the structure and properties of blast furnace slag under different basicity conditions. J. Mol. Liq. 2020, 299, 112195. [Google Scholar] [CrossRef]
- Yu, K.; Zhang, Y.l.; Zhang, S.; Gao, M.; Zhou, C.Q. Effect of Al2O3/TiO2/Na2O component in steelmaking slag on the corrosion of MgO refractories. Ironmak. Steelmak. 2023, 50, 648–657. [Google Scholar] [CrossRef]
- El-Geassy, A.; Shehata, K.; Nasr, M.; Fakhoury, S. Effect of alkalies on the performance of blast furnace. Trans. Iron Steel Inst. Jpn. 1986, 26, 865–874. [Google Scholar] [CrossRef]
- Shiau, J.-S.; Liu, S.-H.; Ho, C.-K. Effect of magnesium and aluminum oxides on fluidity of final blast furnace slag and its application. Mater. Trans. 2012, 53, 1449–1455. [Google Scholar] [CrossRef]
- Vo, D.-H.; Hwang, C.-L.; Yehualaw, M.D.; Liao, M.-C. The influence of MgO addition on the performance of alkali-activated materials with slag−rice husk ash blending. J. Build. Eng. 2021, 33, 101605. [Google Scholar] [CrossRef]
- Jiang, C.; Li, K.; Zhang, J.; Qin, Q.; Liu, Z.; Liang, W.; Sun, M.; Wang, Z. Molecular dynamics simulation on the effect of MgO/Al2O3 ratio on structure and properties of blast furnace slag under different basicity conditions. Metall. Mater. Trans. B 2019, 50, 367–375. [Google Scholar] [CrossRef]
- Zhao, H.; Li, J.; Yang, S.; Liu, J.; Liu, W. Molecular dynamics study of structural properties of refining slag with various CaO/Al2O3 ratios. Minerals 2021, 11, 398. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The nose-hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Le Roux, S.; Petkov, V. ISAACS–interactive structure analysis of amorphous and crystalline systems. Appl. Crystallogr. 2010, 43, 181–185. [Google Scholar] [CrossRef]
- Miyake, A. Interatomic potential parameters for molecular dynamics simulation of crystals in the system K2O–Na2O–CaO–MgO–Al2O3–SiO2. Mineral. J. 1998, 20, 189–194. [Google Scholar] [CrossRef]
- Bale, C.W.; Belisle, E.; Chartrand, P.; Decterov, S.A.; Eriksson, G.; Gheribi, A.E.; Hack, K.; Jung, I.-H.; Kang, Y.-B.; Melançon, J.; et al. Reprint of: FactSage thermochemical software and databases, 2010–2016. Calphad 2016, 55, 1–19. [Google Scholar] [CrossRef]
- Lee, S.; Jung, B.; Lee, N.; Nam, W.; Lee, S.-J.; Yun, Y. Application of FactSage® thermodynamic modeling for predicting the ash transformation with temperatures under partial slagging entrained flow coal gasification condition. Mater. Test. 2018, 60, 163–172. [Google Scholar] [CrossRef]
- van Dyk, J.C.; Waanders, F.B.; Benson, S.A.; Laumb, M.L.; Hack, K. Viscosity predictions of the slag composition of gasified coal, utilizing FactSage equilibrium modelling. Fuel 2009, 88, 67–74. [Google Scholar] [CrossRef]
- Sun, Y.; Tan, M.; Li, T.; Li, J.; Shang, B. Study on the structural properties of refining slags by molecular dynamics with deep learning potential. J. Mol. Liq. 2022, 353, 118787. [Google Scholar] [CrossRef]
- Michalet, X. Mean square displacement analysis of single-particle trajectories with localization error: Brownian motion in an isotropic medium. Phys. Rev. E—Stat. Nonlinear Soft Matter Phys. 2010, 82, 041914. [Google Scholar] [CrossRef]
- Wu, T.; Wang, Q.; Yu, C.; He, S. Structural and viscosity properties of CaO-SiO2-Al2O3-FeO slags based on molecular dynamic simulation. J. Non-Cryst. Solids 2016, 450, 23–31. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SiO2 | CaO | MgO | Al2O3 | Na2O | Total | Melting Point (K) | |
|---|---|---|---|---|---|---|---|
| N-1 | 31.82 | 38.18 | 1.00 | 20.00 | 9.00 | 100 | 1597.8 |
| N-2 | 31.82 | 38.18 | 3.00 | 20.00 | 7.00 | 100 | 1613.5 |
| N-3 | 31.82 | 38.18 | 5.00 | 20.00 | 5.00 | 100 | 1655.6 |
| N-4 | 31.82 | 38.18 | 7.00 | 20.00 | 3.00 | 100 | 1688.3 |
| N-5 | 31.82 | 38.18 | 9.00 | 20.00 | 1.00 | 100 | 1733.6 |
| Ion | z (e) | a (Å) | b (Å) | c (Å3(KJ/mol)1/2) | Ion Pair | D (KJ/mol) | a (1/Å) | r0 (Å) |
|---|---|---|---|---|---|---|---|---|
| Si | 1.92 | 0.5983 | 0.025 | 0.00 | Si-O | 63 | 2 | 1.47 |
| Ca | 0.96 | 1.1425 | 0.042 | 30.74 | Al-O | 50.4 | 2 | 1.58 |
| Mg | 0.96 | 0.9400 | 0.040 | 20.49 | Ca-O | 21 | 2 | 2.20 |
| Al | 1.44 | 0.6758 | 0.030 | 0.00 | Mg-O | 42 | 2 | 1.75 |
| Na | 0.48 | 1.0450 | 0.050 | 20.49 | Na-O | - | - | - |
| O | −0.96 | 1.7700 | 0.138 | 51.23 |
| Si-O | Al-O | Ca-O | Mg-O | Na-O | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | CN | R1 | R2 | CN | R1 | R2 | CN | R1 | R2 | CN | R1 | R2 | CN | |
| N-1 | 1.61 | 4.16 | 4.07 | 1.73 | 4.23 | 4.45 | 2.31 | 4.66 | 7.59 | 2.03 | 4.46 | 5.94 | 2.27 | 4.62 | 7.49 |
| N-2 | 1.61 | 4.17 | 4.06 | 1.73 | 4.24 | 4.42 | 2.31 | 4.67 | 7.57 | 2.03 | 4.46 | 5.89 | 2.27 | 4.59 | 7.50 |
| N-3 | 1.61 | 4.17 | 4.05 | 1.73 | 4.26 | 4.39 | 2.31 | 4.68 | 7.46 | 2.03 | 4.44 | 5.81 | 2.28 | 4.63 | 7.27 |
| N-4 | 1.61 | 4.18 | 4.04 | 1.73 | 4.26 | 4.36 | 2.31 | 4.68 | 7.44 | 2.03 | 4.46 | 5.80 | 2.29 | 4.61 | 7.22 |
| N-5 | 1.61 | 4.18 | 4.04 | 1.73 | 4.27 | 4.32 | 2.31 | 4.69 | 7.42 | 2.03 | 4.46 | 5.72 | 2.31 | 4.63 | 7.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Liu, B.; Zhang, J.; Li, K. Atomistic-Level Insights into MgO and Na2O Modifications of Molten Aluminosilicate Slag: A Molecular Dynamics Research on Structural Evolution and Properties. Metals 2025, 15, 656. https://doi.org/10.3390/met15060656
Jiang C, Liu B, Zhang J, Li K. Atomistic-Level Insights into MgO and Na2O Modifications of Molten Aluminosilicate Slag: A Molecular Dynamics Research on Structural Evolution and Properties. Metals. 2025; 15(6):656. https://doi.org/10.3390/met15060656
Chicago/Turabian StyleJiang, Chunhe, Bo Liu, Jianliang Zhang, and Kejiang Li. 2025. "Atomistic-Level Insights into MgO and Na2O Modifications of Molten Aluminosilicate Slag: A Molecular Dynamics Research on Structural Evolution and Properties" Metals 15, no. 6: 656. https://doi.org/10.3390/met15060656
APA StyleJiang, C., Liu, B., Zhang, J., & Li, K. (2025). Atomistic-Level Insights into MgO and Na2O Modifications of Molten Aluminosilicate Slag: A Molecular Dynamics Research on Structural Evolution and Properties. Metals, 15(6), 656. https://doi.org/10.3390/met15060656