
Effect of Surface Finish on CO2 Corrosion of Low-Alloy Steel in Simulated Sea Water and Well Environments

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Material and Surface Preparation
2.2. Surface Roughness Analysis
2.3. Corrosion Measurements
2.3.1. Local Electrochemical Evaluation Using Scanning Electrochemical Microscopy (SECM)
2.3.2. Immersion Tests and Analysis of Solution Chemistry
2.3.3. Electrochemical Corrosion Measurements
2.4. X-Ray Diffraction and Scanning Electron Microscopy
3. Results
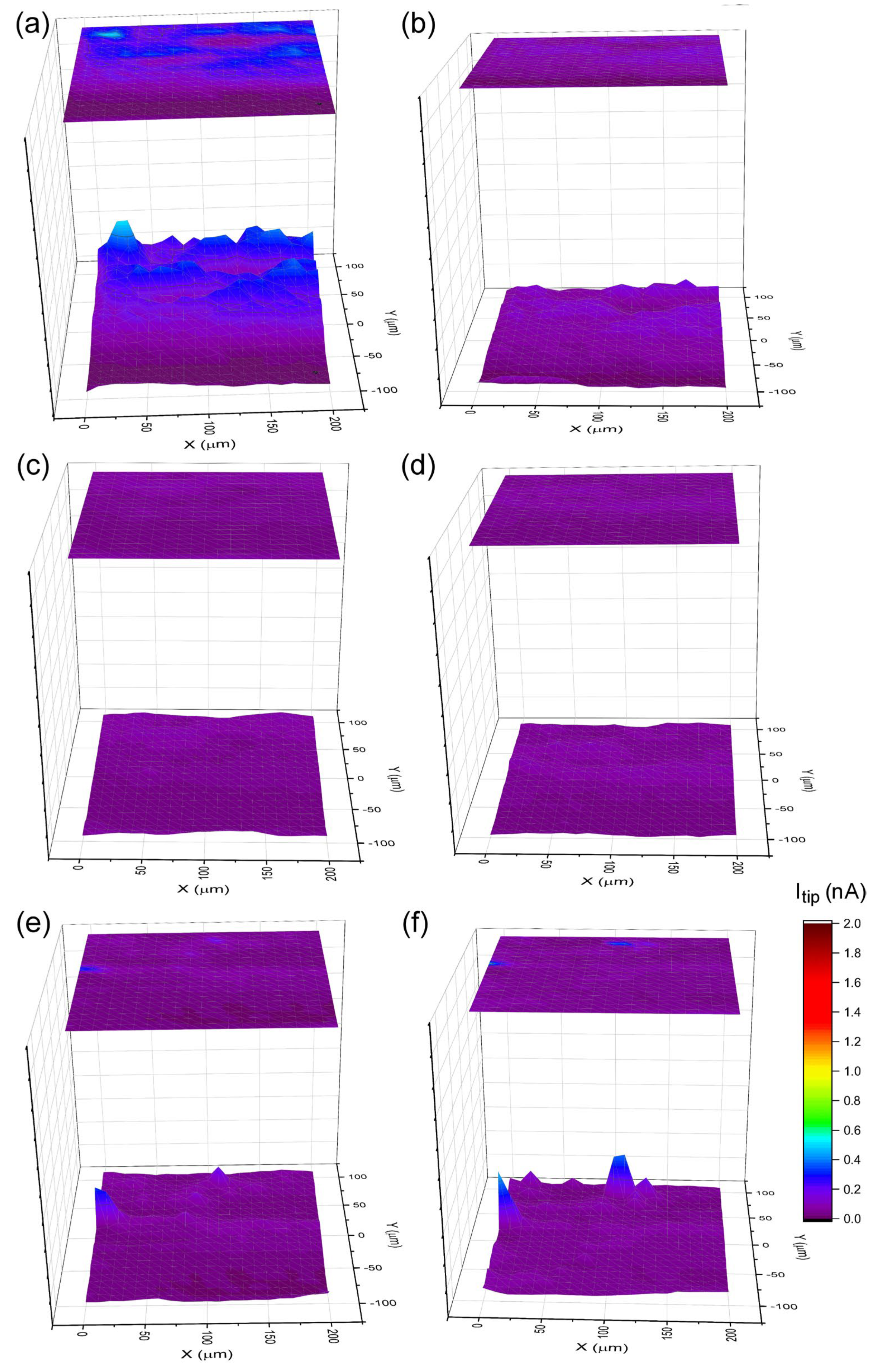
3.1. Surface Characteristics
3.2. Corrosion Investigations
3.2.1. Simulated Sea Water
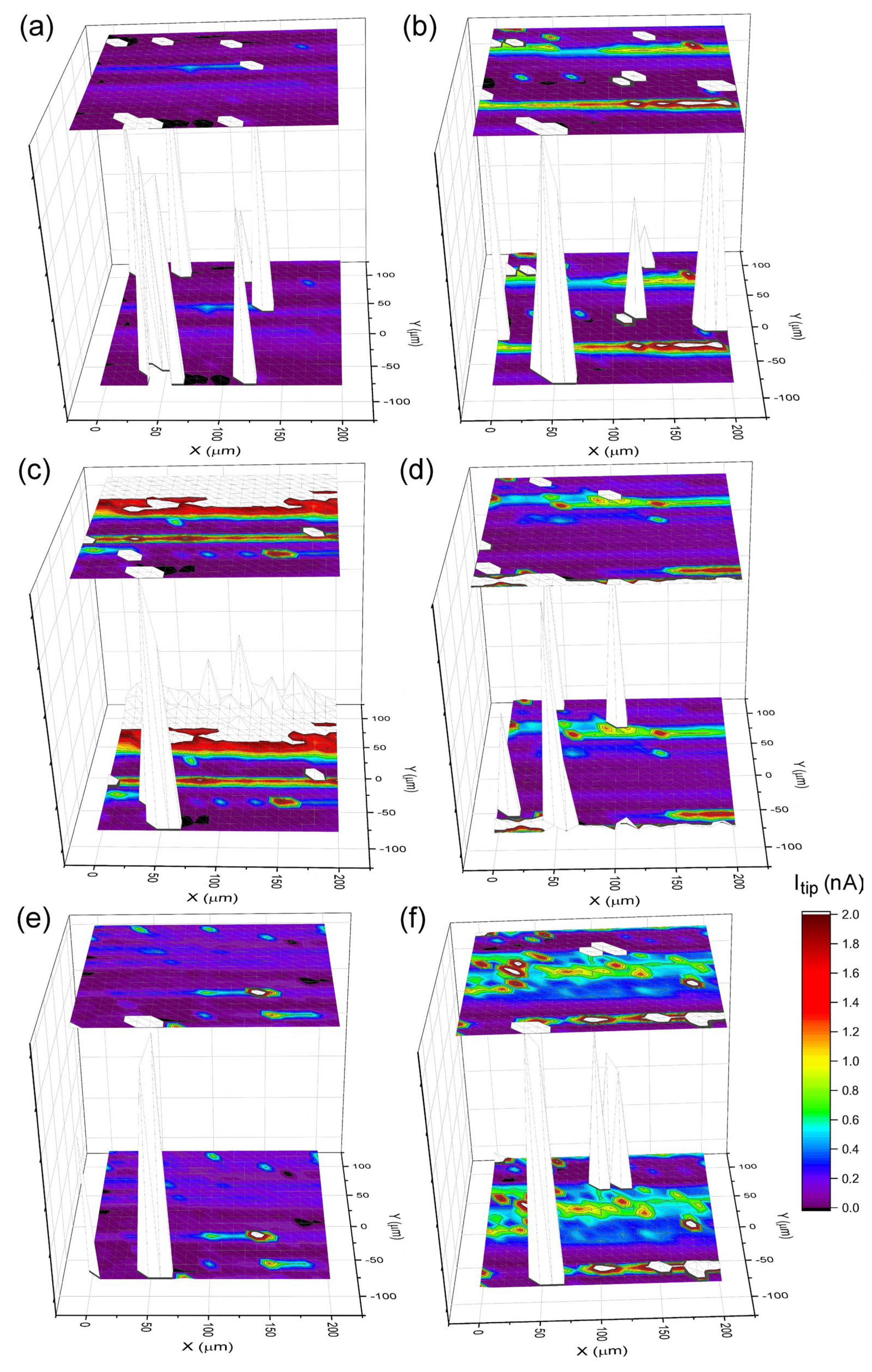
Local Electrochemical Measurements
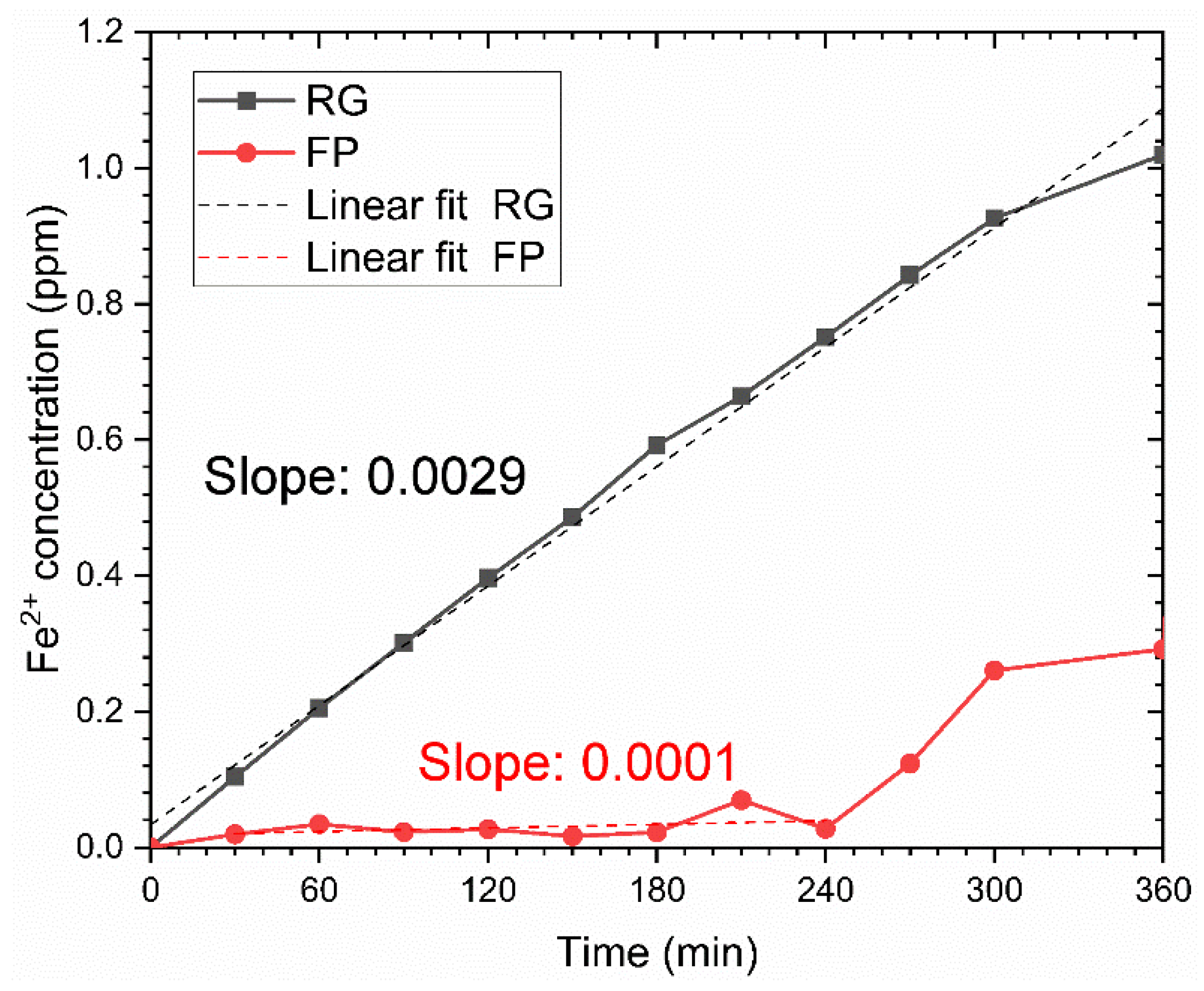
Dissolution Kinetics
3.2.2. Simulated Well Environment
Potentiodynamic Polarization Test
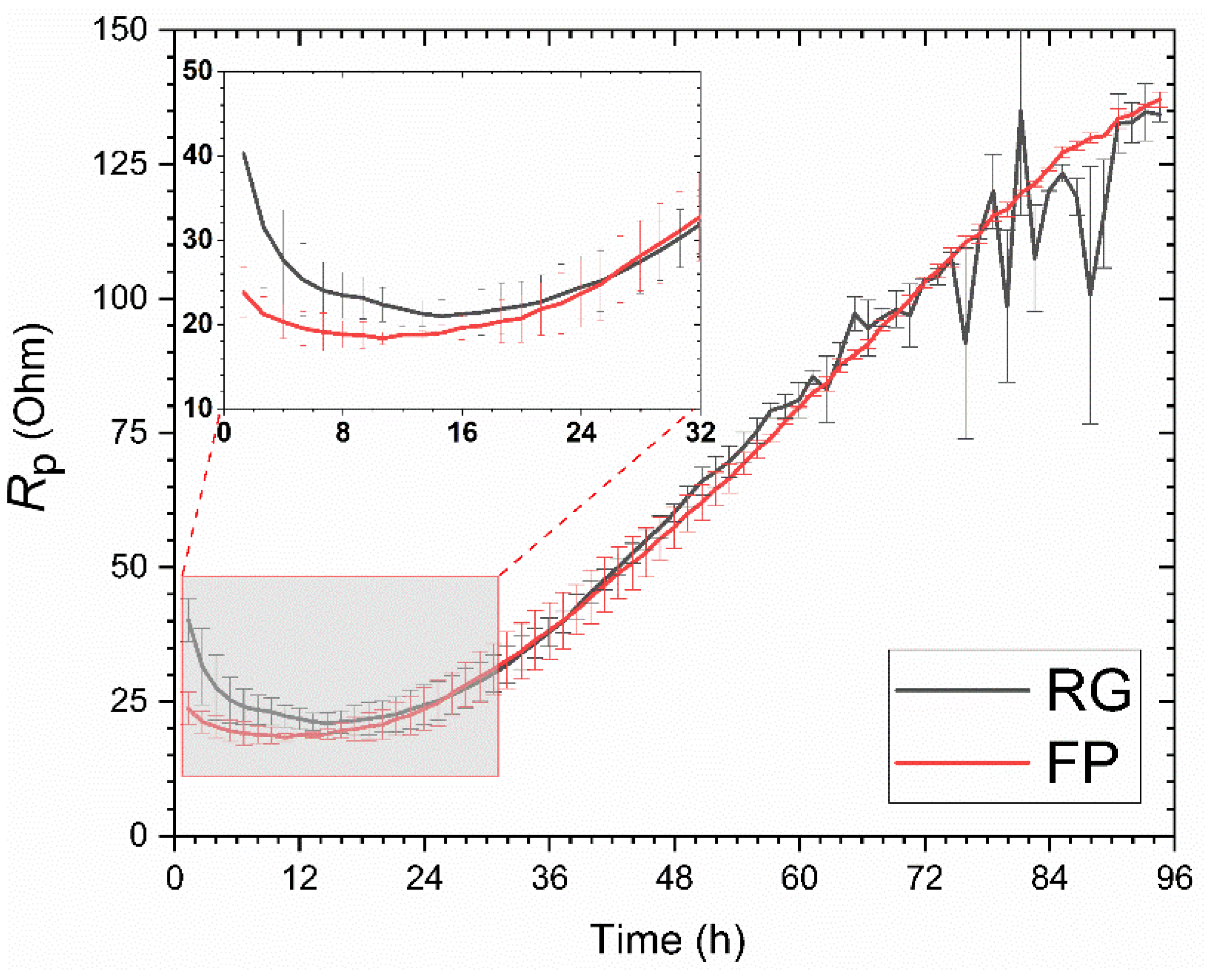
Linear Polarization Resistance Measurements
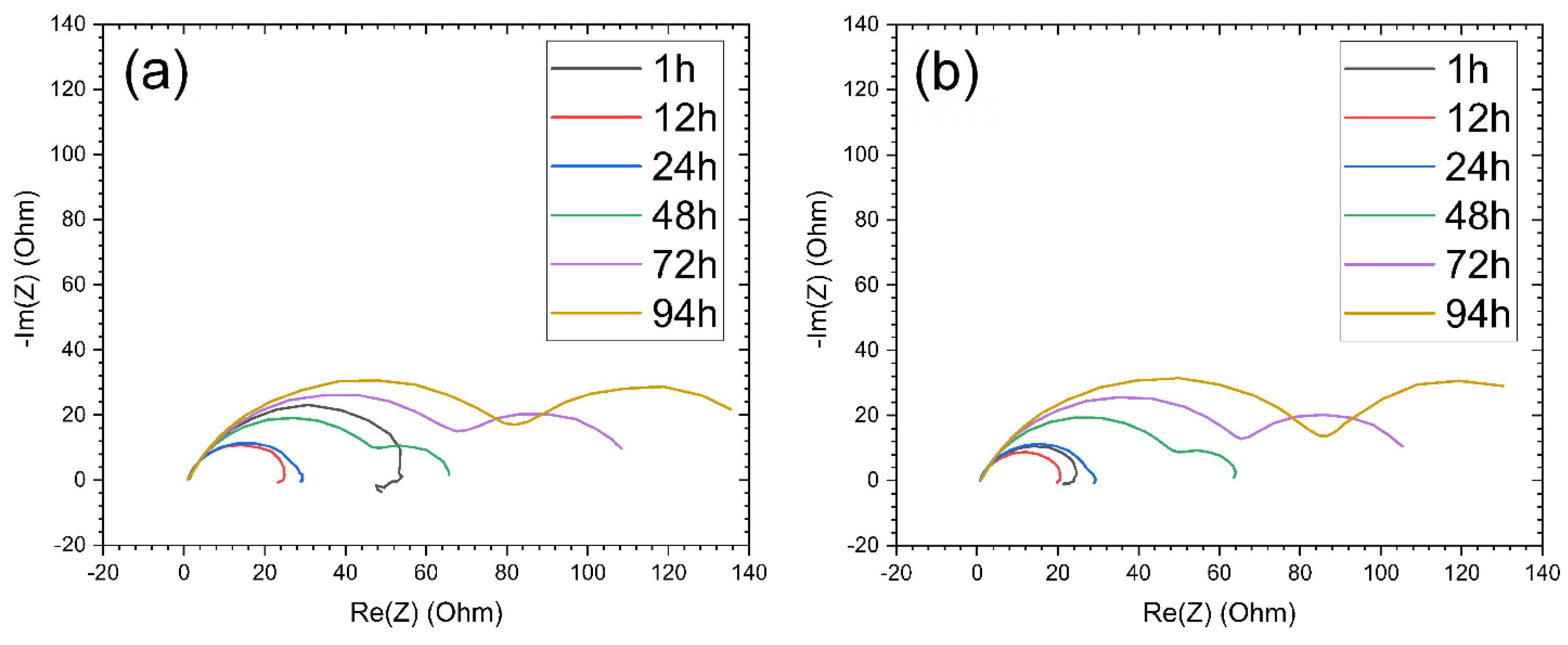
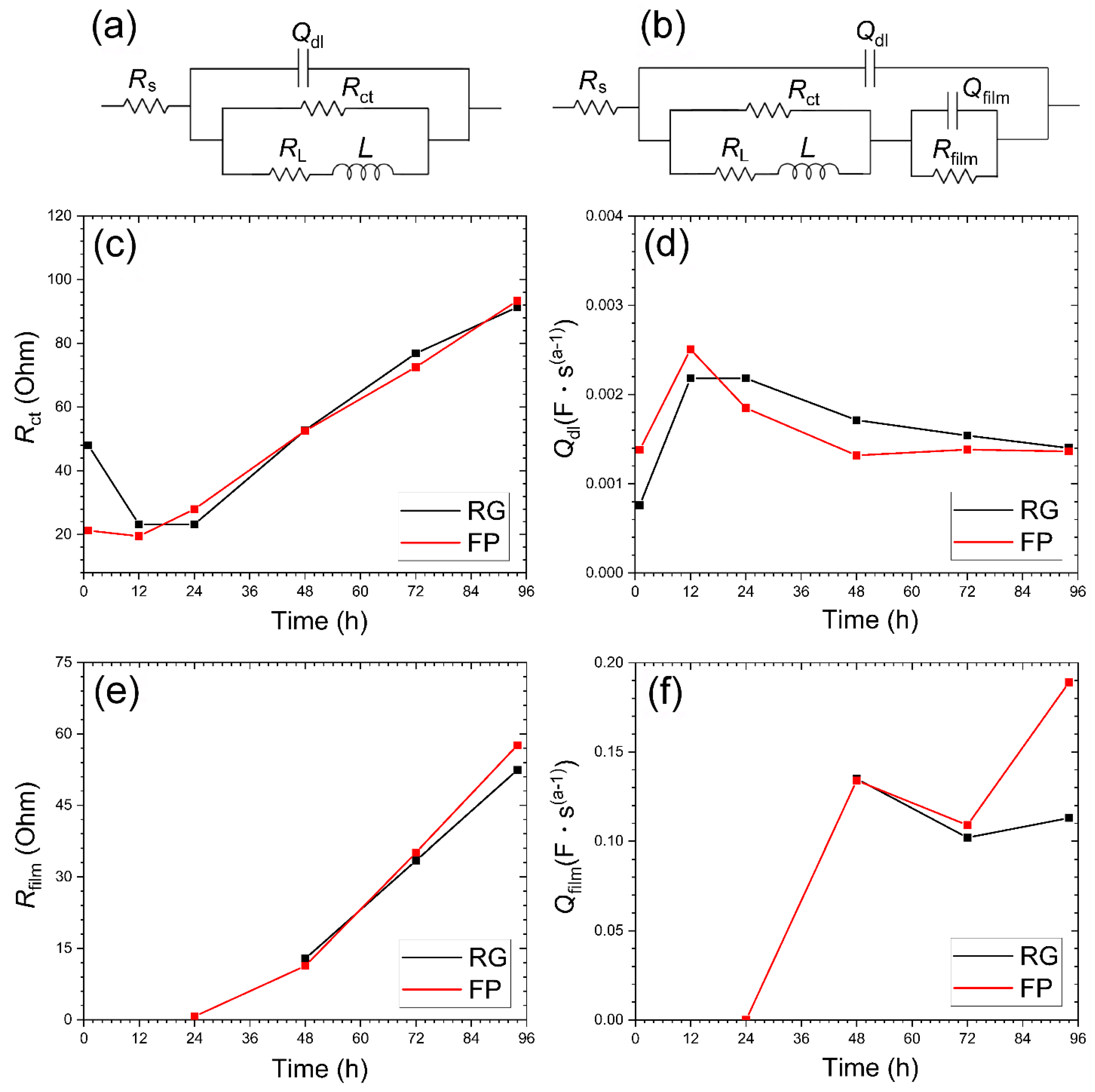
Electrical Impedance Spectroscopy Analysis
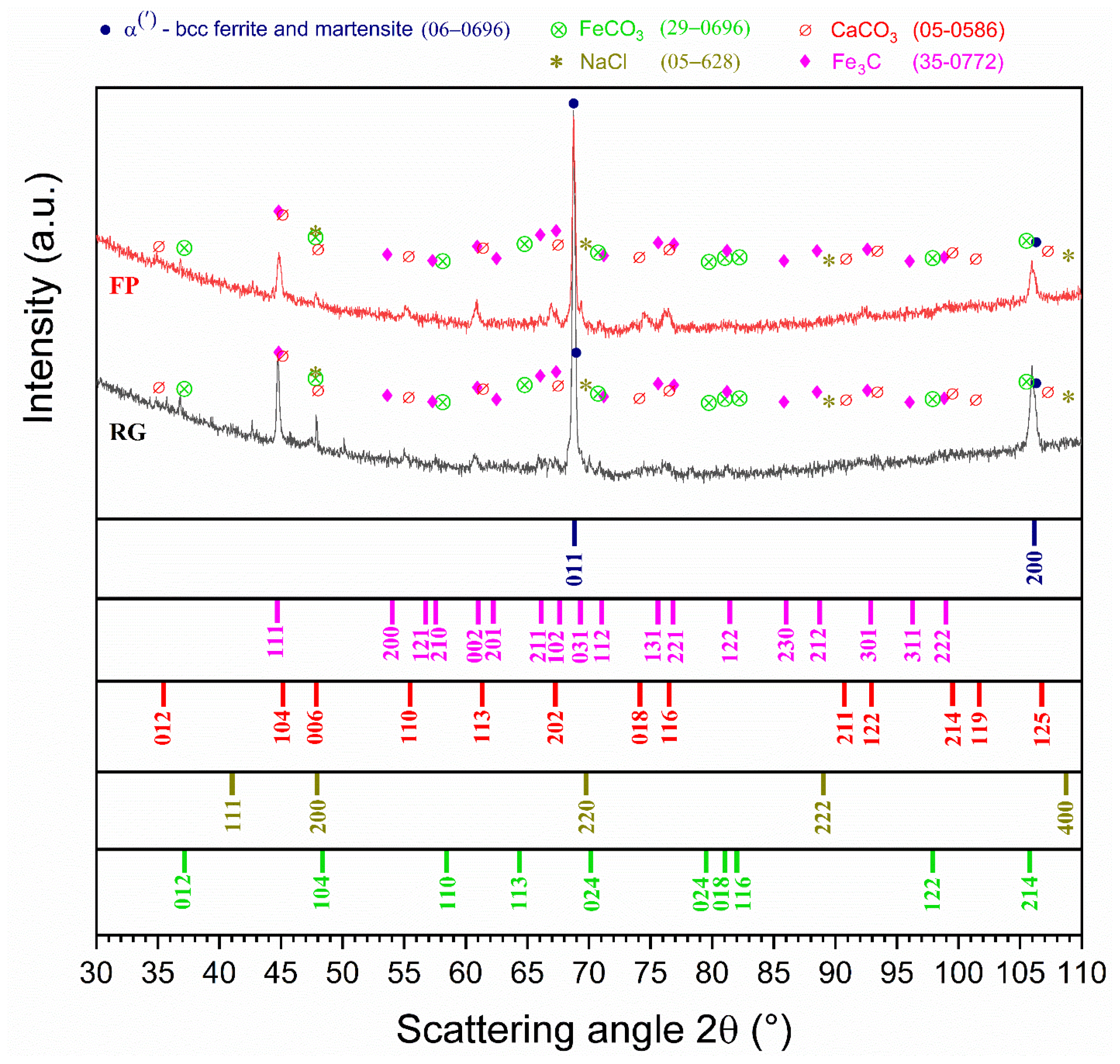
Surface Analysis Using X-Ray Diffraction and SEM-Based Techniques
4. Discussion
5. Conclusions
- The surface finish influences the inherent resistance of the material to CO2 corrosion. The FP sample with a smooth surface exposed to simulated sea water shows higher initial corrosion resistance. On the other hand, the presence of metallic ions in the simulated well environment enables faster precipitation of calcium carbonates, which lowers initially high corrosion rates for the RG sample with a rougher surface.
- The higher rate of corrosion for the RG sample in the simulated well environment allows faster supersaturation of FeCO3 and results in a faster formation of a semi-protective FexCayCO3 scale compared to that of the FP sample. Although the phase composition of the scale is different in the samples with different surface finishes, the influence of the observed differences in the phase composition on corrosion resistance is not significant.
- While the RG sample with the rougher surface shows severe selective corrosion after short-term exposures, the FP sample initially shows slow uniform corrosion, followed by selective corrosion in small areas after longer exposures.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barker, R.; Hua, Y.; Neville, A. Internal corrosion of carbon steel pipelines for dense-phase CO2 transport in carbon capture and storage (CCS)—A review. Int. Mater. Rev. 2017, 62, 1–31. [Google Scholar] [CrossRef]
- Yevtushenko, O.; Bettge, D.; Bohraus, S.; Bäßler, R.; Pfennig, A.; Kranzmann, A. Corrosion behavior of steels for CO2 injection. Process Saf. Environ. Prot. 2014, 92, 108–118. [Google Scholar] [CrossRef]
- Pfennig, A.; Wolf, M.; Kranzmann, A. Corrosion and corrosion fatigue of steels in downhole ccs environment-a summary. Processes 2021, 9, 594. [Google Scholar] [CrossRef]
- Neel, G.; Millet, C.; Greenwood, J.; Rodriguez, D.; Simon, L.; Lima, L. Material Selection for Anthropogenic CO2 Injection, Mechanical and Corrosion Behavior of Steels Under CO2 with Impurities. In Proceedings of the 16th International Conference on Greenhouse Gas Control Technologies, GHGT-16, Lyon, France, 23–27 October 2022; pp. 1–19. [Google Scholar] [CrossRef]
- Kane, R.D. Corrosion in Petroleum Production Operations. In ASM Handbook: Corrosion: Environments and Industries; ASM International: Almere, The Netherlands, 2006; pp. 922–966. [Google Scholar] [CrossRef]
- Popoola, L.T.; Grema, A.S.; Latinwo, G.K.; Gutti, B.; Balogun, A.S. Corrosion problems during oil and gas production and its mitigation. Int. J. Ind. Chem. 2013, 4, 35. [Google Scholar] [CrossRef]
- Moiseeva, L.S. Carbon dioxide corrosion of oil and gas field equipment. Prot. Met. 2005, 41, 76–83. [Google Scholar] [CrossRef]
- Kermani, M.B.; Morshed, A. Carbon Dioxide Corrosion in Oil and Gas Production—A Compendium. Corrosion 2003, 59, 659–683. [Google Scholar] [CrossRef]
- Dwivedi, D.; Lepková, K.; Becker, T. Carbon steel corrosion: A review of key surface properties and characterization methods. RSC Adv. 2017, 7, 4580–4610. [Google Scholar] [CrossRef]
- Gupta, K.K.; Haratian, S.; Mishin, O.V.; Ambat, R. CO2 corrosion resistance of low-alloy steel tempered at different temperatures. Corros. Sci. 2024, 232, 112027. [Google Scholar] [CrossRef]
- Nešić, S. Key issues related to modelling of internal corrosion of oil and gas pipelines—A review. Corros. Sci. 2007, 49, 4308–4338. [Google Scholar] [CrossRef]
- Li, W.; Xiong, Y.; Brown, B.; Kee, K.E.; Nesic, S. Measurement of wall shear stress in multiphase flow and its effect on protective FeCO3 corrosion product layer removal. In Proceedings of the CORROSION 2015, Dallas, TX, USA, 15–19 March 2015; pp. 1–15. [Google Scholar]
- Tanupabrungsun, T.; Brown, B.; Nesic, S. Effect of pH on CO2 Corrosion of Mild Steel at Elevated Temperatures. In Proceedings of the CORROSION 2013, Orlando, FL, USA, 17–21 March 2015; pp. 1–11. [Google Scholar]
- López, D.A.; Pérez, T.; Simison, S.N. The influence of microstructure and chemical composition of carbon and low alloy steels in CO2 corrosion. A state-of-the-art appraisal. Mater. Des. 2003, 24, 561–575. [Google Scholar] [CrossRef]
- Gupta, K.K.; Haratian, S.; Gupta, S.; Mishin, O.V.; Ambat, R. The effect of cooling rate-induced microstructural changes on CO2 corrosion of low alloy steel. Corros. Sci. 2022, 209, 110769. [Google Scholar] [CrossRef]
- Gupta, K.K.; Yazdi, R.; Styrk-Geisler, M.; Mishin, O.V.; Ambat, R. Effect of microstructure of low-alloy steel on corrosion propagation in a simulated CO2 environment. J. Electrochem. Soc. 2022, 169, 111504. [Google Scholar] [CrossRef]
- Gupta, K.K.; Haratian, S.; Mishin, O.V.; Ambat, R. The impact of minor Cr additions in low alloy steel on corrosion behavior in simulated well environment. Npj Mater. Degrad. 2023, 7, 1–12. [Google Scholar] [CrossRef]
- Haratian, S.; Gupta, K.K.; Larsson, A.; Abbondanza, G.; Bartawi, E.H.; Carlà, F.; Lundgren, E.; Ambat, R. Ex-situ synchrotron X-ray diffraction study of CO2 corrosion-induced surface scales developed in low-alloy steel with different initial microstructure. Corros. Sci. 2023, 222, 111387. [Google Scholar] [CrossRef]
- Poulson, B. Mass transfer from rough surfaces. Corros. Sci. 1990, 30, 743–746. [Google Scholar] [CrossRef]
- Tantirige, S.; Trass, O. Mass transfer at geometrically dissimilar rough surfaces. Can. J. Chem. Eng. 1984, 62, 490–496. [Google Scholar] [CrossRef]
- Postlethwaite, J.; Lotz, U. Mass transfer at erosion corrosion roughened surfaces. Can. J. Chem. Eng. 1988, 66, 75–78. [Google Scholar] [CrossRef]
- Cornet, I.; Lewis, W.; Kappesser, R. Effect of surface roughness on mass transfer to a rotating disc. Trans. Inst. Chem. Eng. 1969, 47, 222–226. [Google Scholar]
- Gabe, D.; Makanjuola, P. Enhanced mass transfer using roughened rotating cylinder electrodes in turbulent flow. J. Appl. Electrochem. 1987, 17, 370–384. [Google Scholar] [CrossRef]
- Al-Khateeb, M.; Barker, R.; Neville, A.; Thompson, H. An experimental and theoretical investigation of the influence of surface roughness on corrosion in CO2 environments. J. Corros. Sci. Eng. 2017, 20, 1–29. [Google Scholar]
- King, C.V.; Howard, P.L. Heat Transfer and Diffusion Rates at Solid- Liquid Boundaries. Ind. Eng. Chem. 1937, 29, 75–78. [Google Scholar] [CrossRef]
- Brennan, W.C. Mass Transfer in Turbulent Pipe Flow. The Effect of Surface Roughness. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 1963. [Google Scholar]
- Fogg, G.; Morse, J. Development of a new solvent-free flow efficiency coating for natural gas pipelines. In Proceedings of the Rio Pipeline Conference & Exposition 2005, IBP1233, Rio de Janeiro, Brazil, 17–19 October 2005. [Google Scholar]
- Nordsveen, M.; Nešić, S.; Nyborg, R.; Stangeland, A. A Mechanistic Model for Carbon Dioxide Corrosion of Mild Steel in the Presence of Protective Iron Carbonate Films—Part 1: Theory and Verification. Corrosion 2003, 59, 443–456. [Google Scholar] [CrossRef]
- Asma, R.B.A.N.; Yuli, P.A.; Mokhtar, C.I. Study on the effect of surface finish on corrosion of carbon steel in CO2 environment. J. Appl. Sci. 2011, 11, 2053–2057. [Google Scholar] [CrossRef]
- Nesic, S.; Postlethwaite, J.; Thevenot, N. Superposition of diffusion and chemical reaction controlled limiting currents—Application to CO2 corrosion. J. Corros. Sci. Eng. 1995, 1, 1–14. [Google Scholar]
- Mahato, B.K.; Voora, S.K.; Shemilt, L.W. Steel pipe corrosion under flowconditions-I. An isothermal correlation for a mass transfer model. Corros. Sci. 1968, 8, 173–180. [Google Scholar] [CrossRef]
- Ma, W.; Wang, H.; Wang, Y.; Neville, A.; Hua, Y. Experimental and simulation studies of the effect of surface roughness on corrosion behaviour of X65 carbon steel under intermittent oil/water wetting. Corros. Sci. 2022, 195, 109947. [Google Scholar] [CrossRef]
- Silverman, D.C. The rotating cylinder electrode for examining velocity-sensitive corrosion—A review. Corrosion 2004, 60, 1003–1023. [Google Scholar] [CrossRef]
- Li, H.; Li, D.; Zhang, L.; Bai, Y.; Wang, Y.; Lu, M. Fundamental aspects of the corrosion of N80 steel in a formation water system under high CO2 partial pressure at 100 °C. RSC Adv. 2019, 9, 11641–11648. [Google Scholar] [CrossRef]
- Rizzo, R.; Ambat, R. Effect of initial CaCO3 saturation levels on the CO2 corrosion of 1Cr carbon steel. Mater. Corros. 2021, 72, 1076–1090. [Google Scholar] [CrossRef]
- Bai, H.; Cui, X.; Wang, R.; Lv, N.; Yang, X.; Li, R.; Ma, Y. Effect of Surface Roughness on Static Corrosion Behavior of J55 Carbon Steel in CO2-Containing Geothermal Water at 65 °C. Coatings 2023, 13, 821. [Google Scholar] [CrossRef]
- Al-Khateeb, M.A.M.J. An Experimental and Modelling Study of The Effect of Surface Roughness on Mass Transfer and Corrosion in CO2 Saturated Oilfield Environments, The University of Leeds. 2019. Available online: http://etheses.whiterose.ac.uk/24279/ (accessed on 28 July 2022).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | Cr | Mn | Mo | P | Si | S | Ni | Cu | Fe |
|---|---|---|---|---|---|---|---|---|---|
| 0.40 | - | 1.9 | - | 0.03 | 0.45 | 0.030 | 0.25 | 0.35 | Bal. |
| Unit | Na+ | Cl- | Ca2+ | Fe2+ | |
|---|---|---|---|---|---|
| Mol | 1.13 | 1.43 | 0.15 | 0.015 | 0.0016 |
| Ppm | 27,450 | 53,000 | 6012 | 917 | 90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, K.K.; Pedroni, S.; Mercier, A.; Haratian, S.; Mishin, O.V.; Ambat, R. Effect of Surface Finish on CO2 Corrosion of Low-Alloy Steel in Simulated Sea Water and Well Environments. Metals 2025, 15, 302. https://doi.org/10.3390/met15030302
Gupta KK, Pedroni S, Mercier A, Haratian S, Mishin OV, Ambat R. Effect of Surface Finish on CO2 Corrosion of Low-Alloy Steel in Simulated Sea Water and Well Environments. Metals. 2025; 15(3):302. https://doi.org/10.3390/met15030302
Chicago/Turabian StyleGupta, Kapil Kumar, Sarah Pedroni, Alexia Mercier, Saber Haratian, Oleg V. Mishin, and Rajan Ambat. 2025. "Effect of Surface Finish on CO2 Corrosion of Low-Alloy Steel in Simulated Sea Water and Well Environments" Metals 15, no. 3: 302. https://doi.org/10.3390/met15030302
APA StyleGupta, K. K., Pedroni, S., Mercier, A., Haratian, S., Mishin, O. V., & Ambat, R. (2025). Effect of Surface Finish on CO2 Corrosion of Low-Alloy Steel in Simulated Sea Water and Well Environments. Metals, 15(3), 302. https://doi.org/10.3390/met15030302