
Corrosion Behavior of X65 API 5L Carbon Steel Under Simulated Storage Conditions: Influence of Gas Mixtures, Redox States, and Temperature Assessed Using Electrochemical Methods for up to 100 Hours

 , and
, and
Abstract
1. Introduction
1.1. General Context
1.2. Influence of the Main Gas and the Temperature on the Corrosion of Carbon Steel
2. Materials and Methods
2.1. Cox Pore Water Characteristics and Reconstitution of a Basic Pore Water Before Its Equilibration with Three Different Gas Mixtures
2.1.1. Cox Pore Water (CPW)
2.1.2. Basic Ionic Reconstitution of CPW (Or IRCPW) and Its Equilibration with Its Natural Gas Mixture
2.1.3. Modus Operandi for the Reconstitution of the Three Representative Corrosive Media for the Present Study
2.1.4. Modelling of the Three Corrosive Media at Two Temperatures
2.2. Carbon Steel API 5L X65 Characteristics and Electrodes Designed for Corrosion Studies
2.3. Experimental Setup for Studying the Corrosivity of AM-IRCPW, BM-IRCPW, and CM-IRCPW Waters Against CS-X65
2.3.1. The Electrochemical Reactor
- A refrigerant, into which water flows at 1 °C, was used to condense vapors and minimize losses of AM-IRCPW, BM-IRCPW, and CM-IRCPW waters.
- Six essential electrodes were used to monitor the physical and chemical parameters of the fluid as well as to investigate the reactivity of CS-X65:
- ◦
- A combined pH glass electrode (InLab Reach, Mettler Toledo, Columbus, OH, USA) that was systematically calibrated between each experiment using commercial standard pH buffer solutions (4 and 7);
- ◦
- A platinum wire electrode was used to monitor the Platinum open circuit potential (OCPPt or redox potential) versus the internal reference of the pH electrode;
- ◦
- An electrochemical triplet was used only for electrochemical corrosion measurement. It included a CS-X65 working electrode (WE) and a saturated calomel reference electrode (SCE), which consisted of a commercial SCE (K0077 from protected with a KCl 3 mol L−1 junction K0065 both from AMETEK, Inc. Berwyn, PA, USA) and a 6 cm heigh cylindrical Pt/Ir grid counter electrode (CE) with a diameter of 6 cm. The SCE is at approximately +240 mV/SHE;
- ◦
- A CS-X65, called the free electrode, is used to monitor its OCPX65 without external electrochemical disturbances;
- ◦
- A Pyrex® tube glass bubbler comprising a dip tube and a diffuser is used for gas equilibration using humidified gas mixtures. The reactor was filled with 1 L of deaerated (humidified N2, 99.99999%, 1 bar) prospective IRCPW continuously stirred. It was then equilibrated using one of the three humidified gas mixtures before temperature was raised to the desired values at 25 °C or 80 °C.

2.3.2. Electrochemical Apparatuses, pH Meter, Data Logger
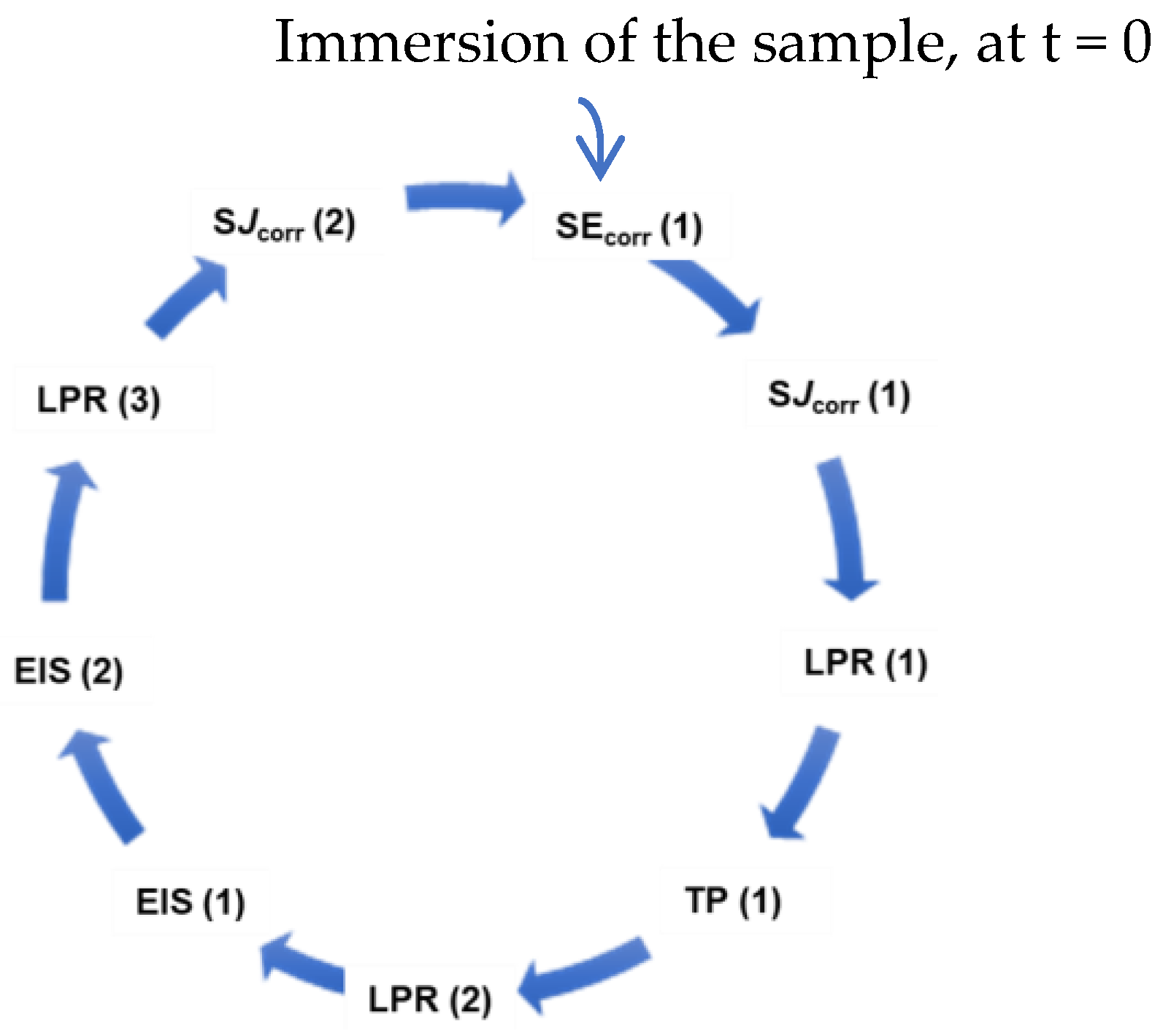
2.3.3. Electrochemical Techniques
2.3.4. Gravimetric or Mass Loss Technique
2.3.5. Electrochemical Study
- -
- SEcorr: monitoring of the free corrosion potential Ecorr;
- -
- SJcorr: monitoring of the corrosion-free current Jcorr to the corrosion-free potential Ecorr;
- -
- LPR: linear polarization resistance measurement, allowing access to Jcorr;
- -
- TP: linear polarization (drawing of the Tafel curves);
- -
- EIS: measurement of impedance by EIS at Ecorr.
2.4. Scanning Electron Microscopy (SEM) Characterization
3. Results
3.1. pH and EPt Temporal Evolutions Under the Three Conditions at Two Temperatures
3.1.1. pH Temporal Evolutions
3.1.2. EPt Temporal Evolutions
3.2. EX65 Temporal Evolutions Under the Three Conditions at Two Temperatures
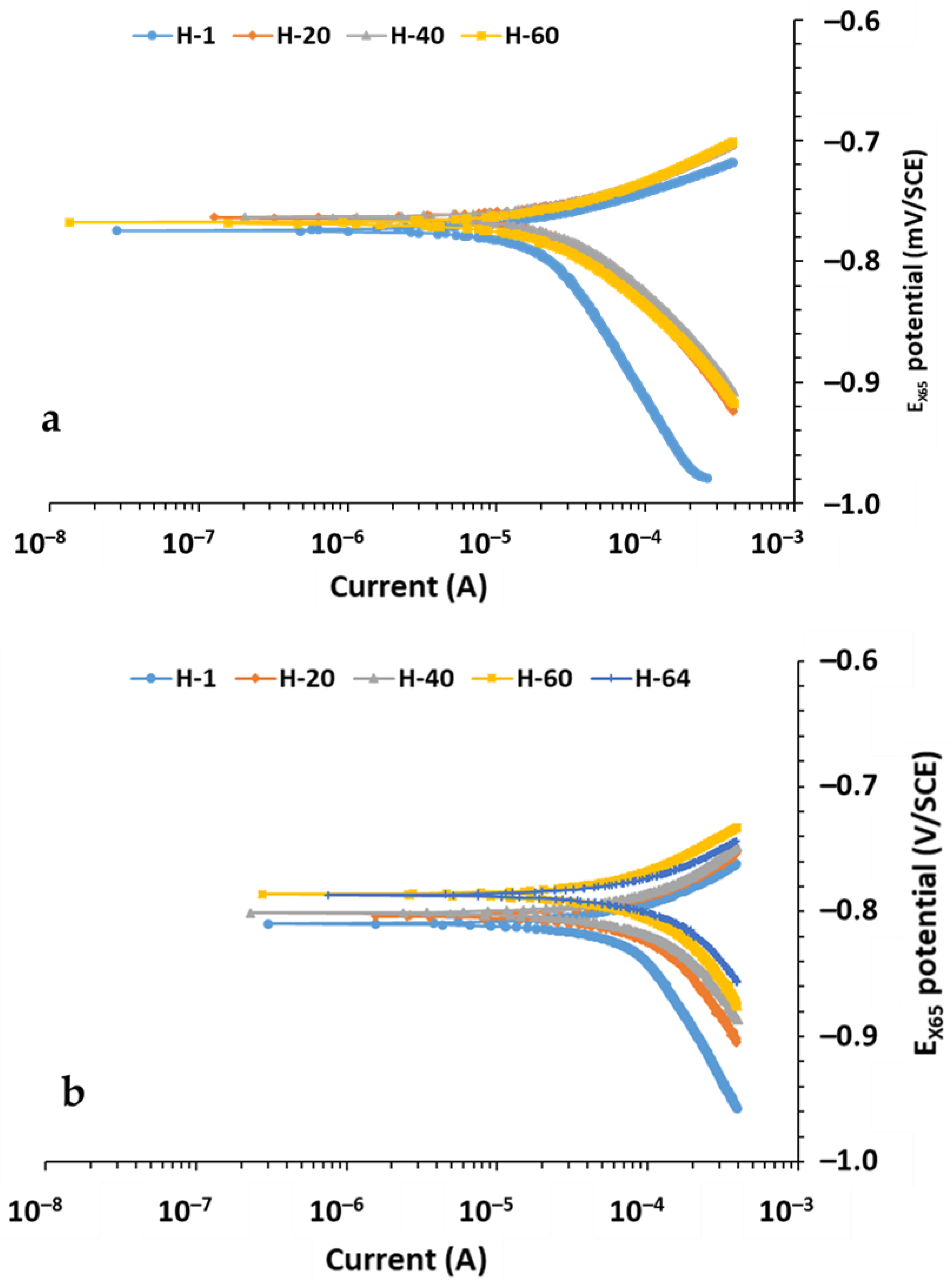
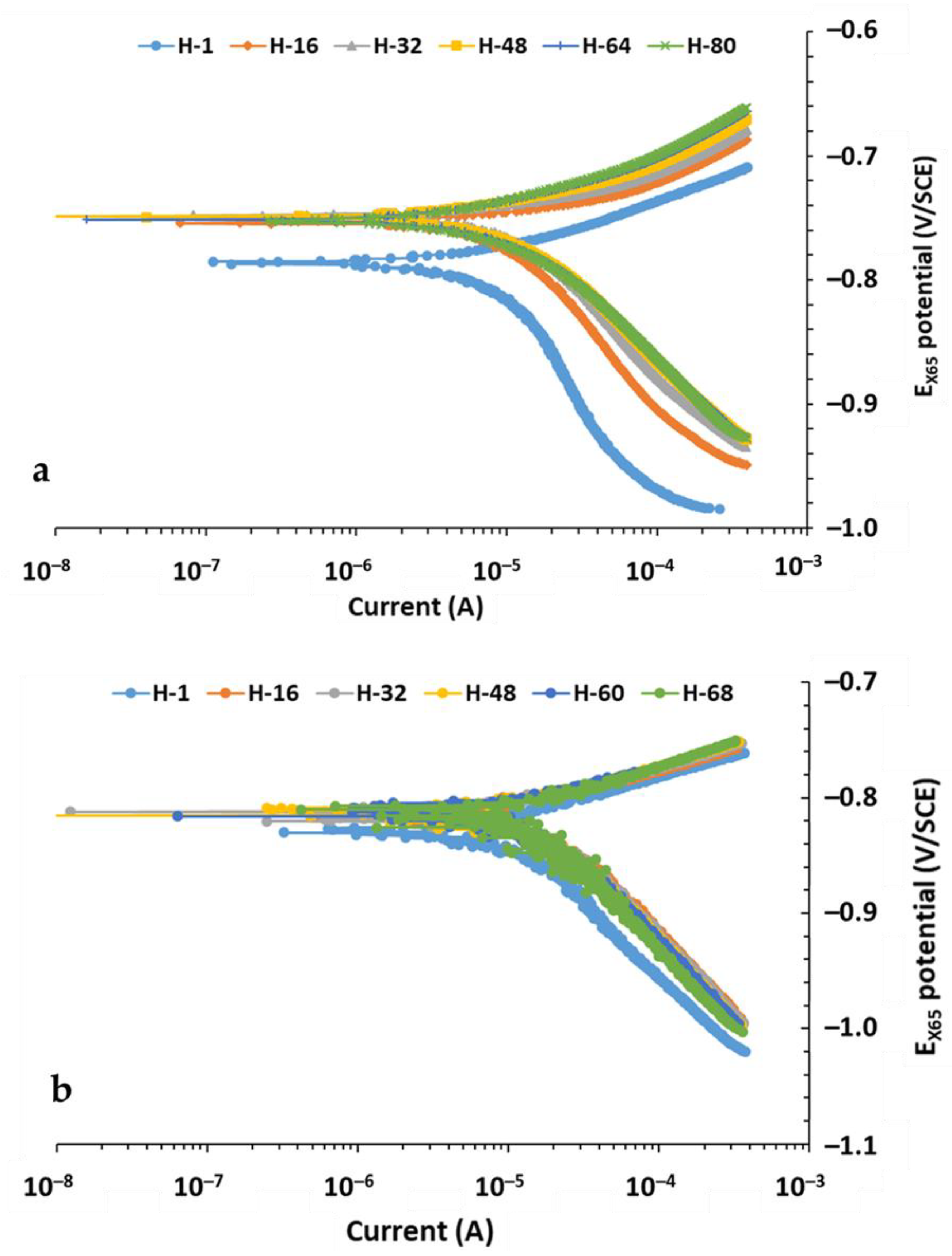
3.3. Tafel Results Under the Three Conditions at Two Temperatures
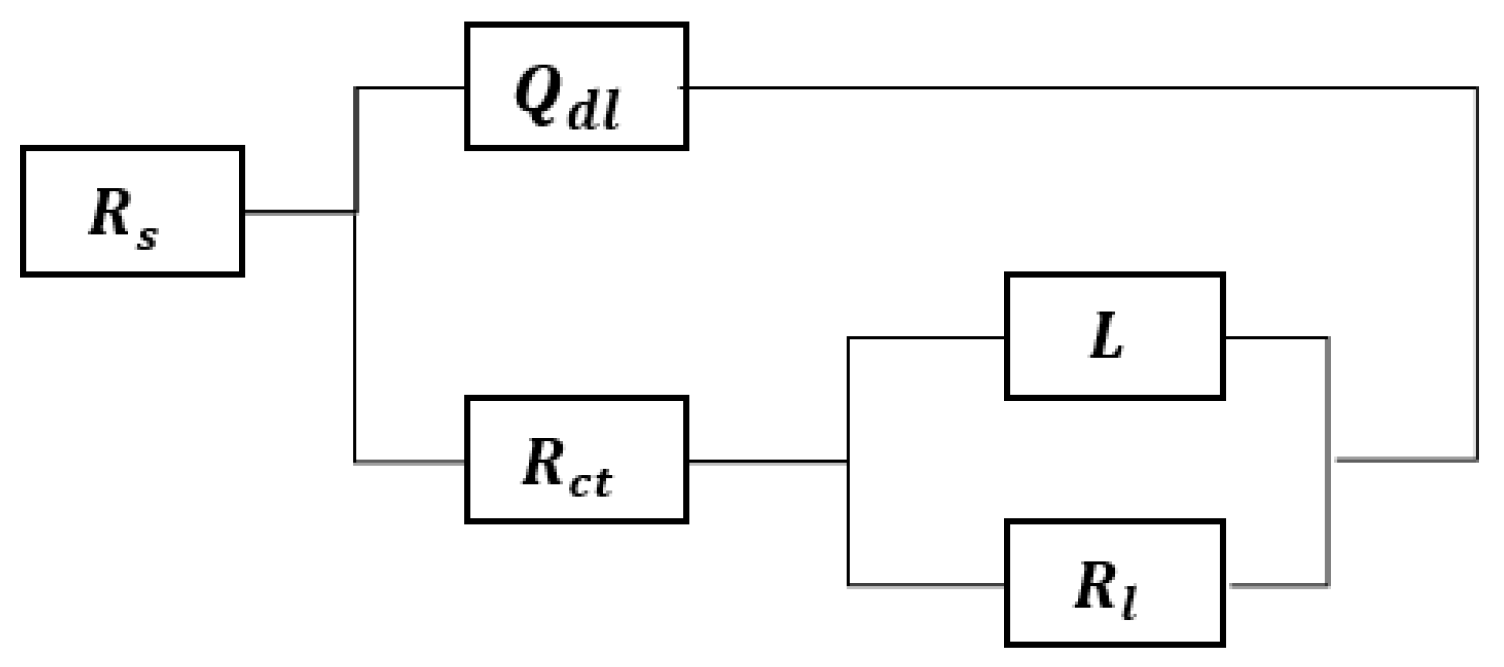
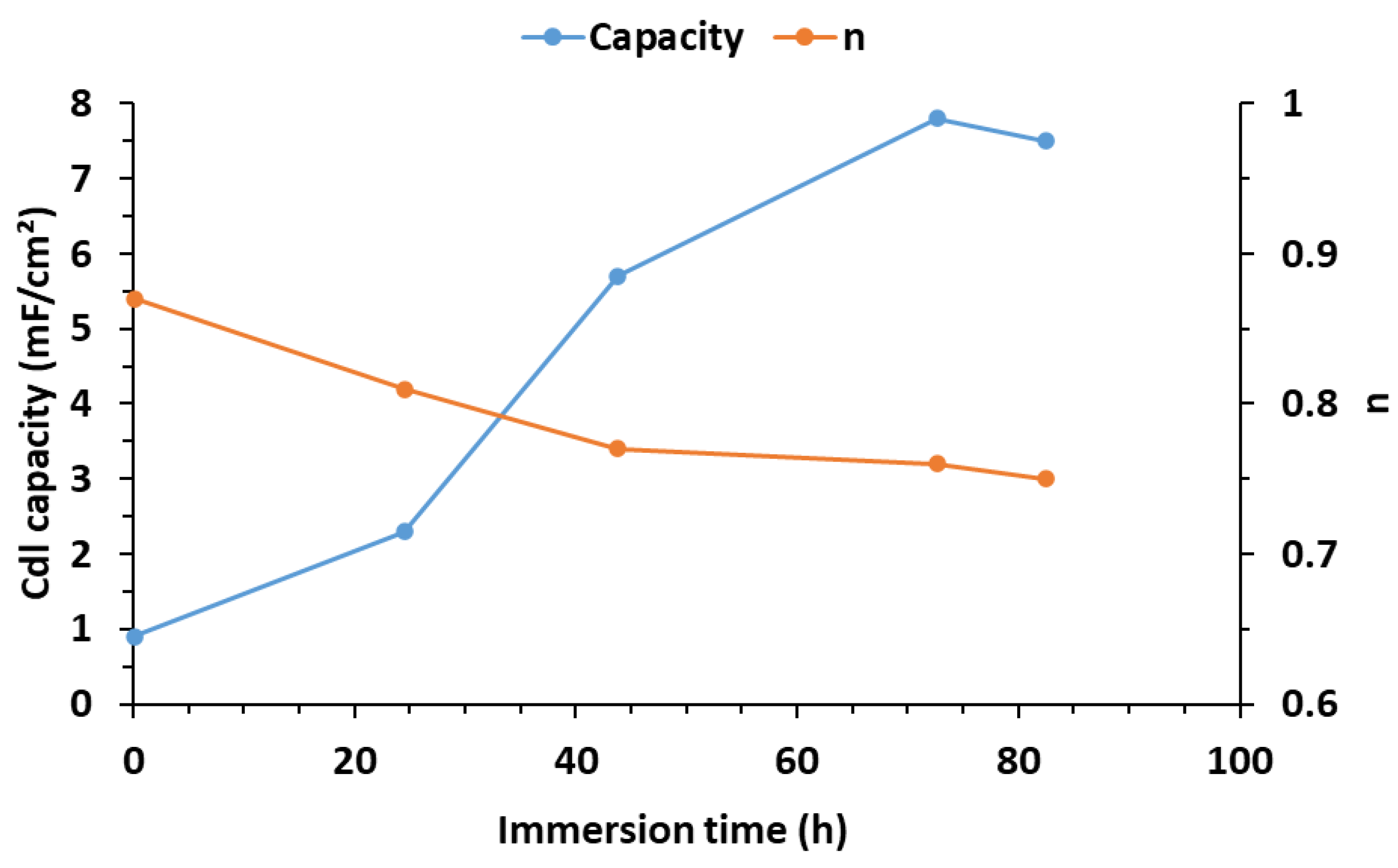
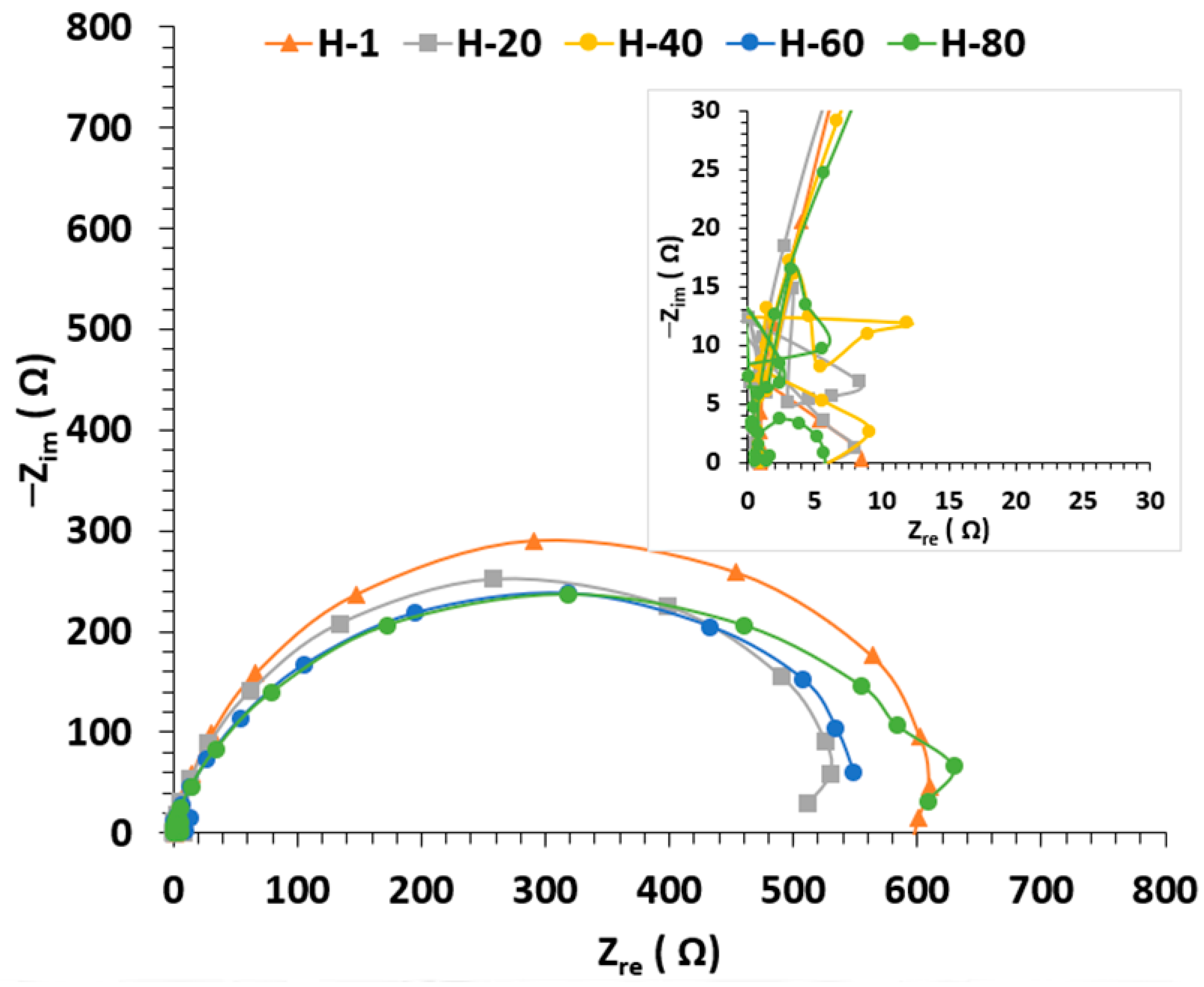
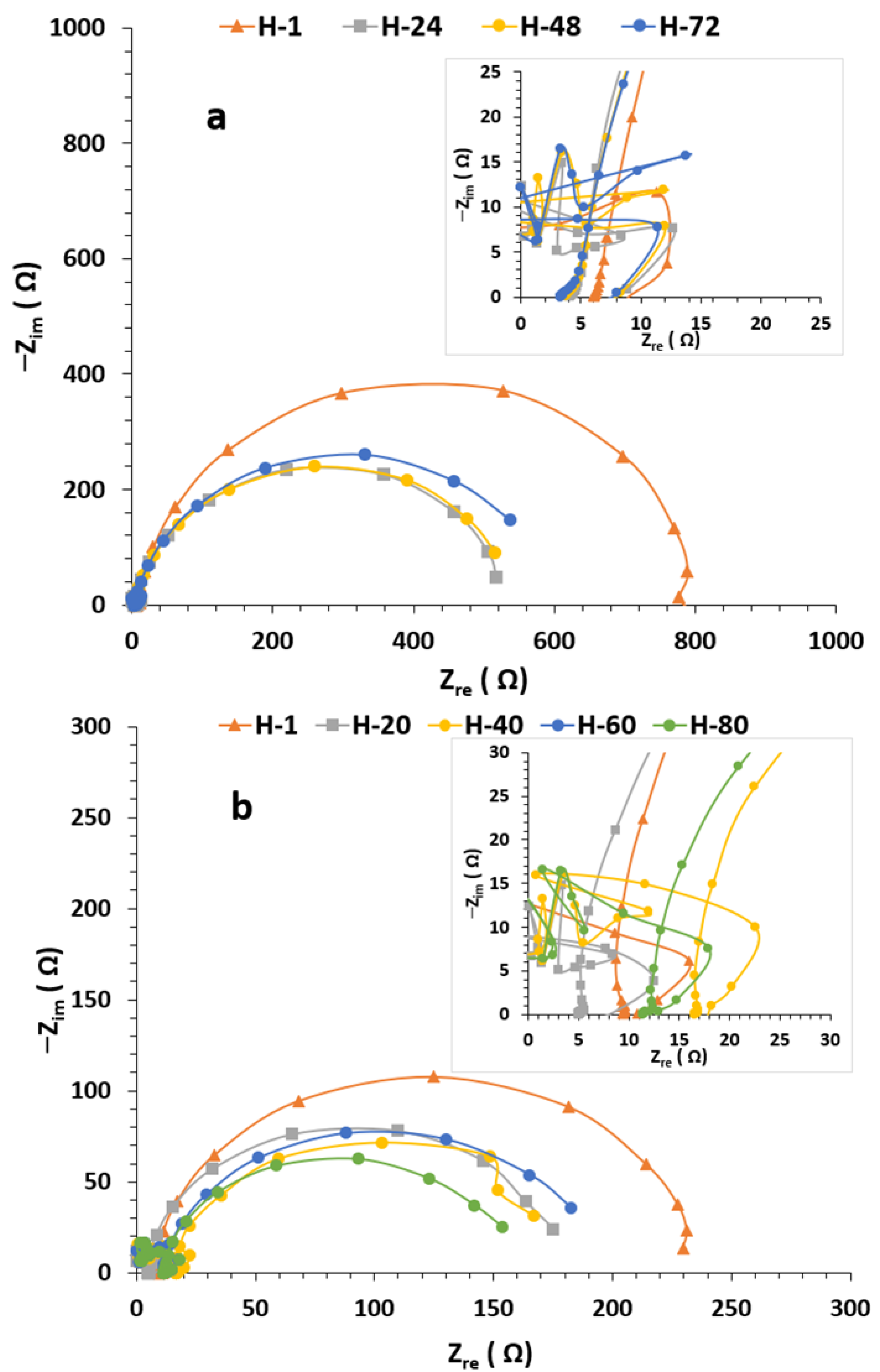
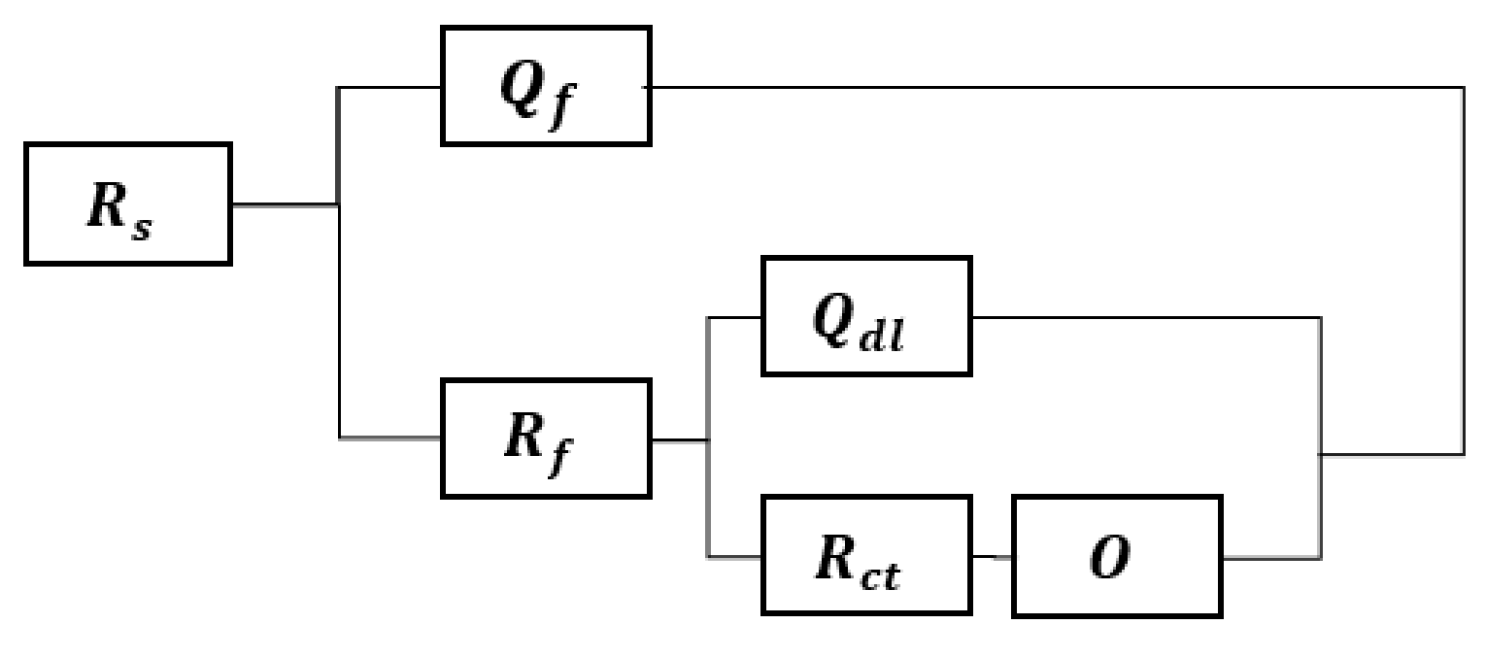
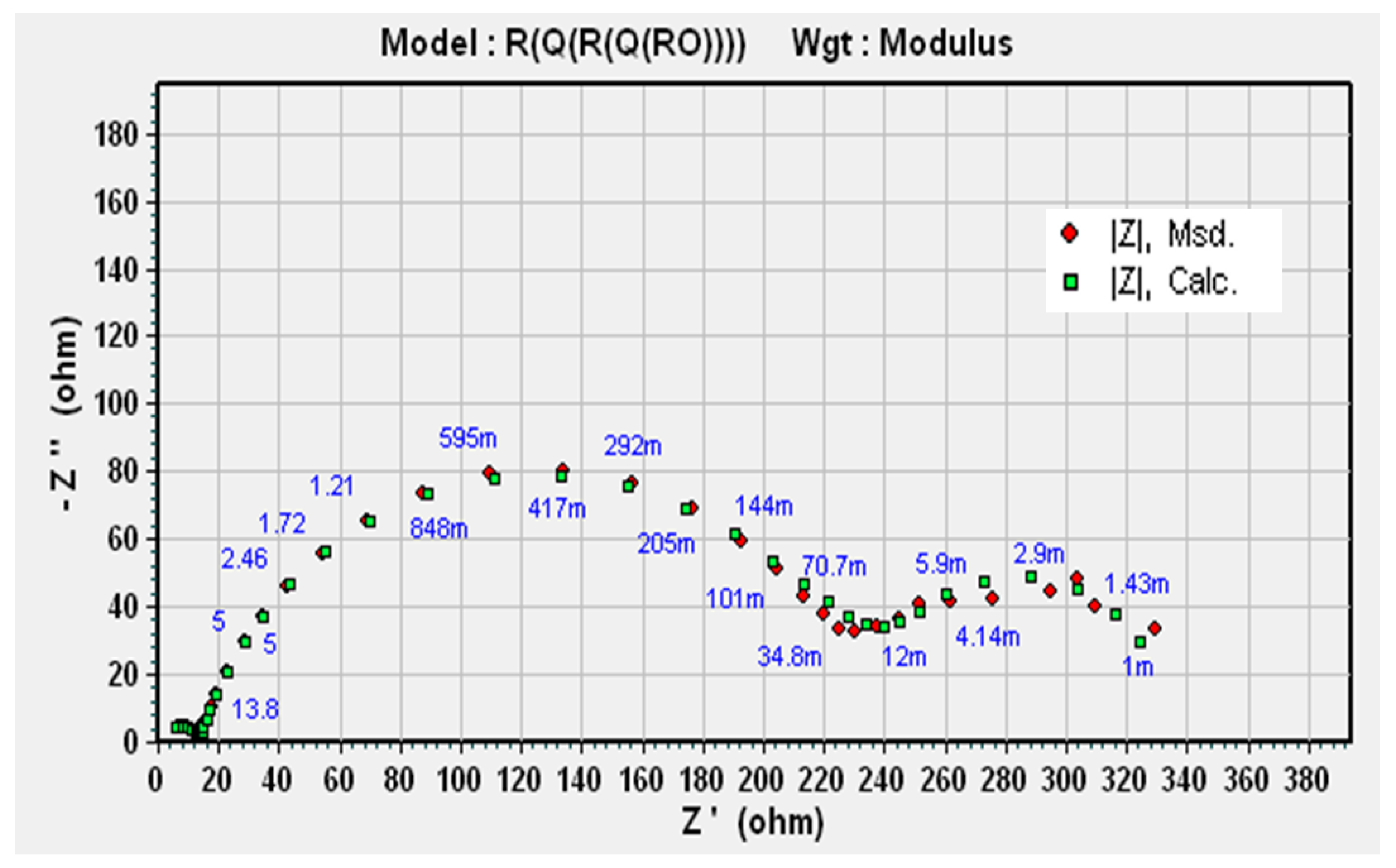
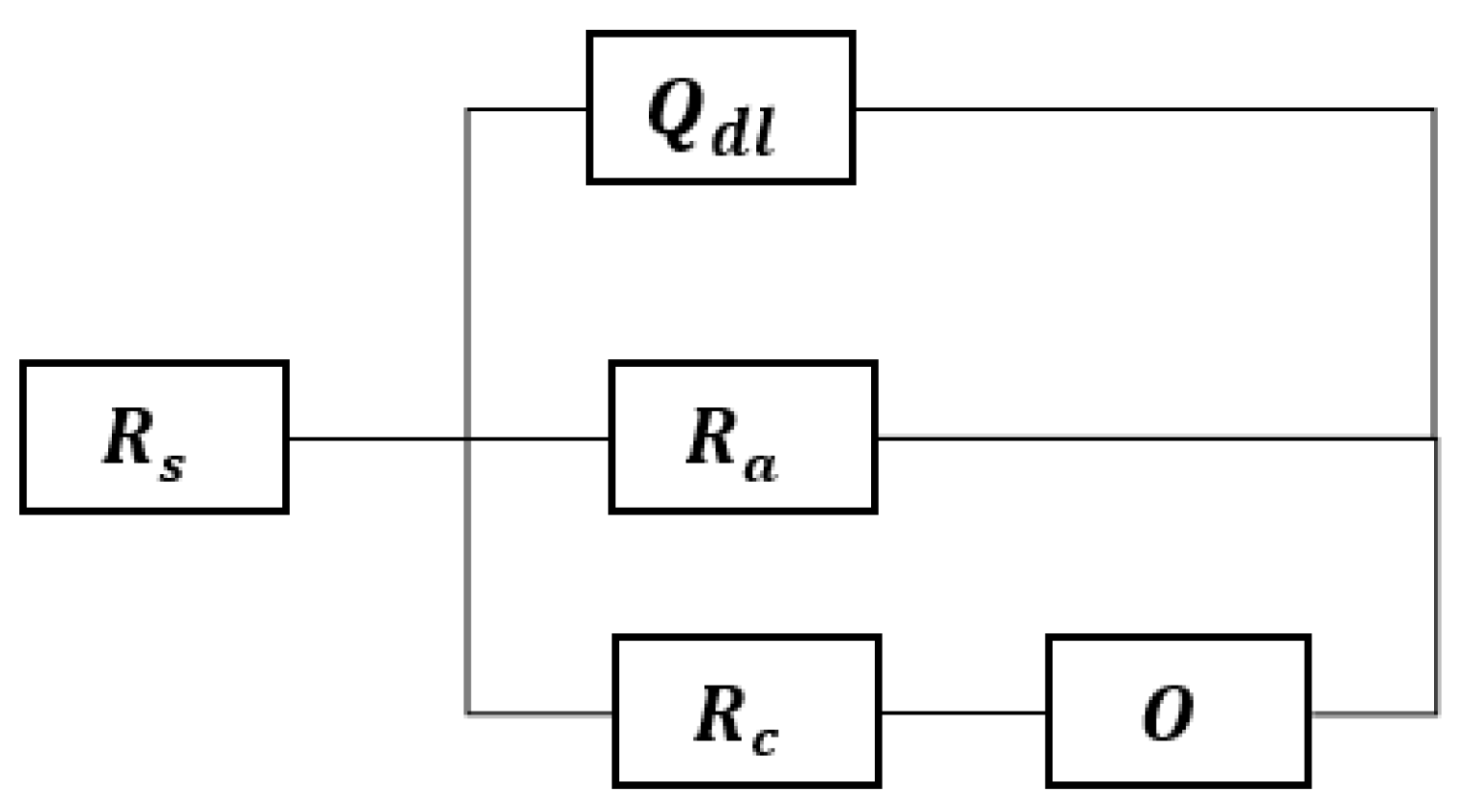
3.4. EIS Results
3.4.1. Condition A (CO2 1%, N2 99%)
3.4.2. Condition B (H2S 0.8%, CO2 20%, N2 79.2%)
3.4.3. Condition C (O2 20%, N2 80%)
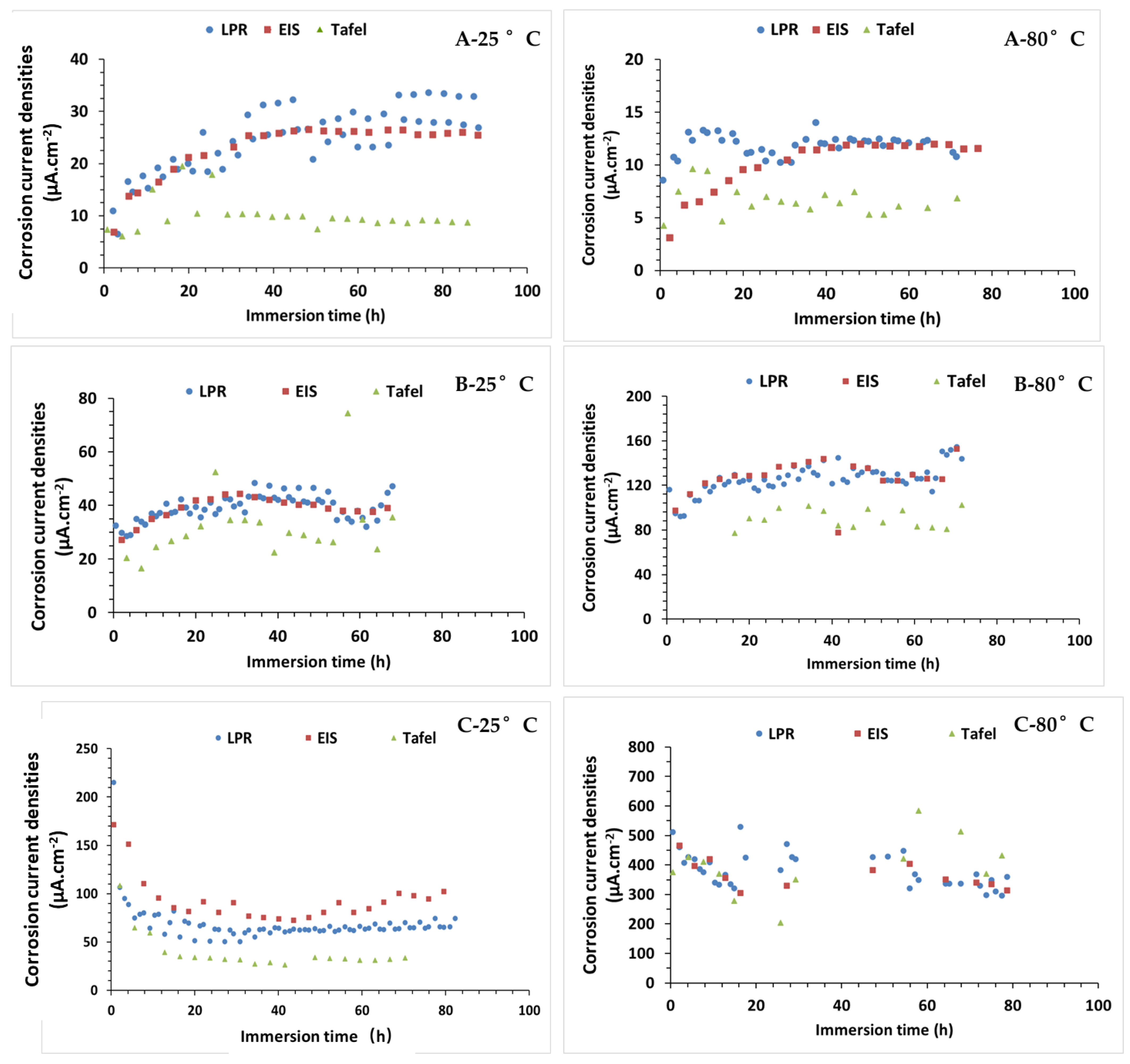
3.5. Corrosion Current Densities (CCD)
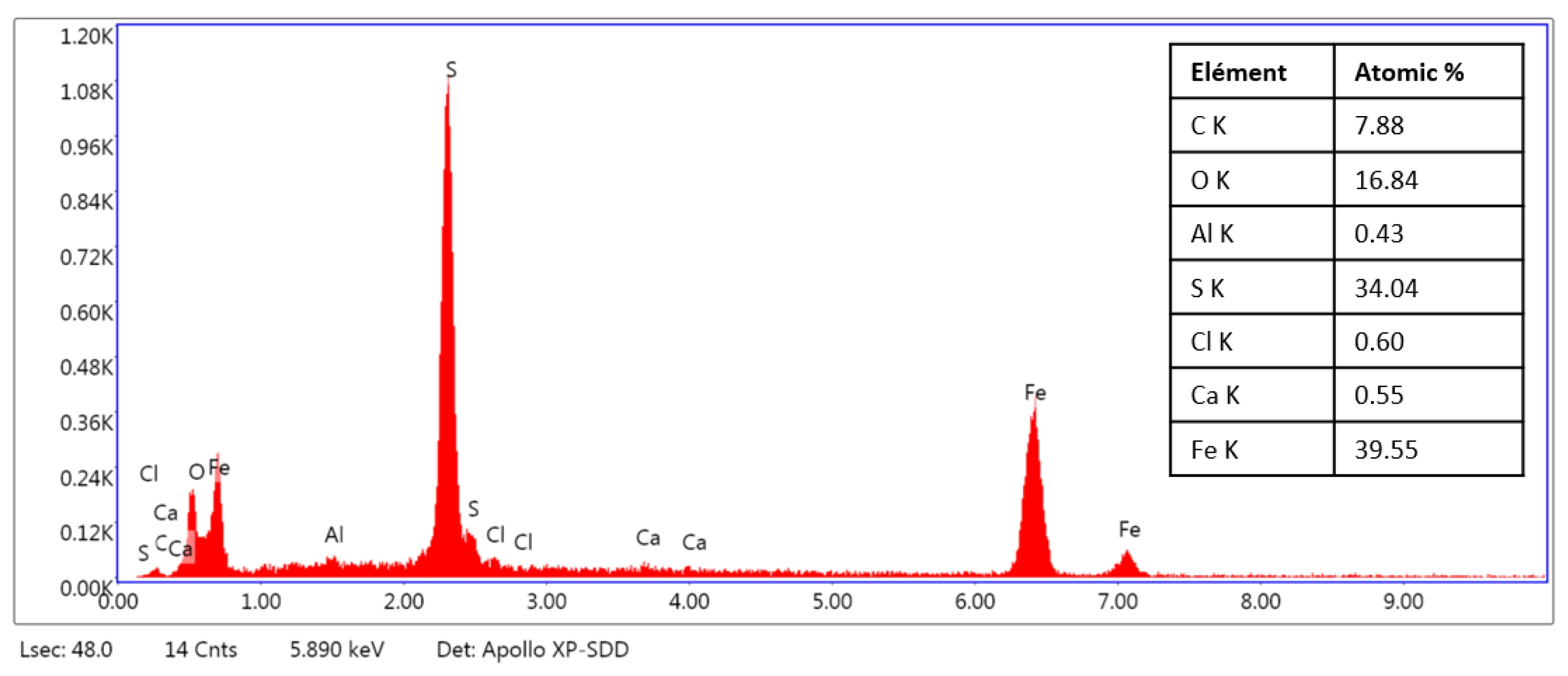
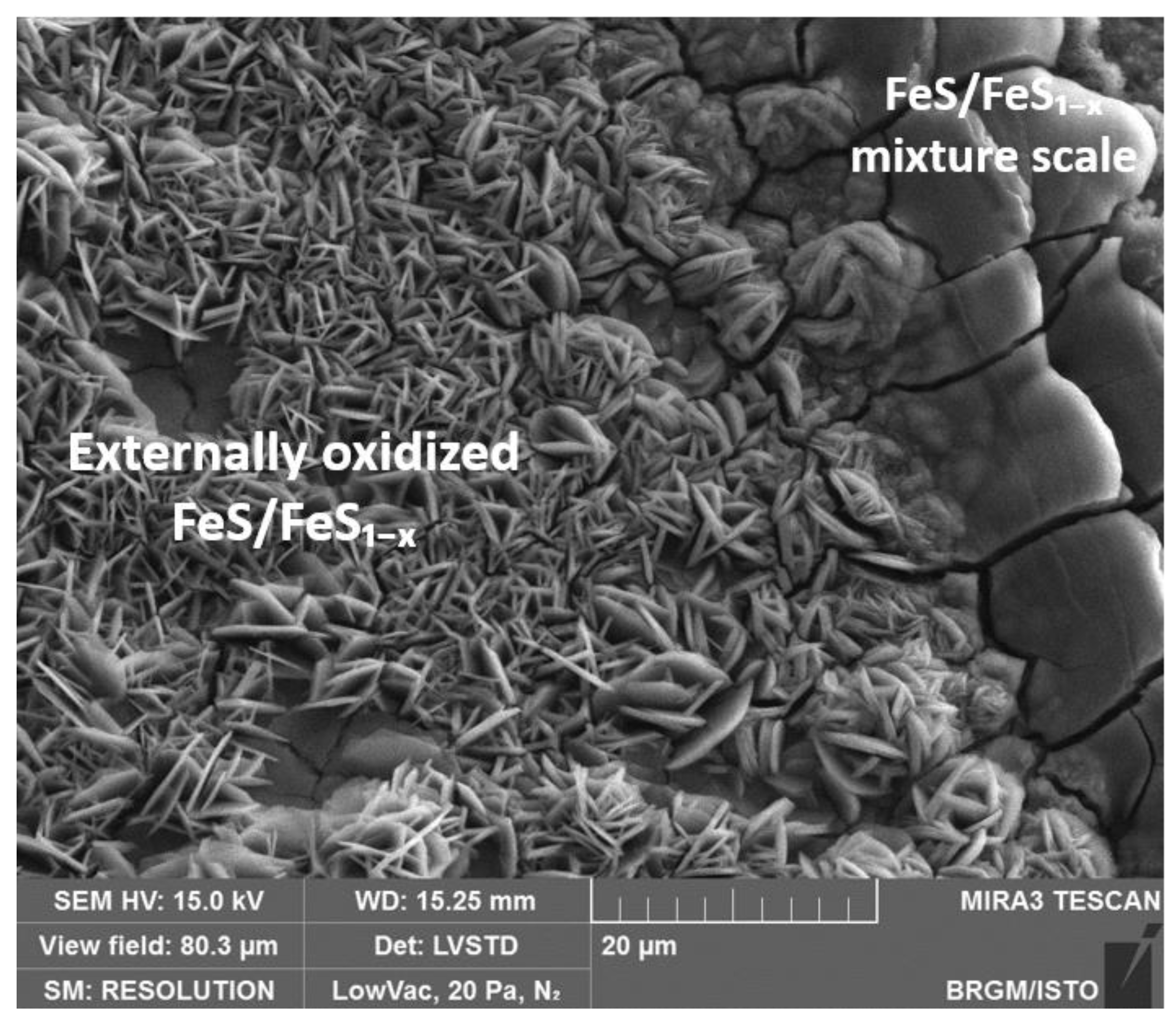
3.6. SEM Characterization of the CS-X65 Surface After Immersion in BM-IRCPW (0.8% H2S, 20% CO2, 79.2% N2) at 80 °C
4. Discussion
- -
- The first stage involves the initiation of the steel corrosion with co-adsorption of hydrogen, chloride, and bicarbonate atoms from the first hours of immersion of the steel. The adsorption is clearly visible at low frequencies on the Nyquist plots.
- -
- The second stage involves the formation of deposits of non-crystallized iron carbonates on the surface of the steel at 25 °C and especially at 80 °C (see Table A1 and Table A4). These rather protective deposits do not completely cover the surface of the steel and have the particularity of polarizing the steel. EIS also highlights this based on the appearance in high frequencies of a new interface after a couple of hours.
- -
- The last stage corresponds to the progressive crystallization of iron carbonate deposits into siderite (FeCO3) at 80 °C. This change in the structure of the deposits leads to a relative ennoblement of the carbon steel over time. The polarization resistances at 80 °C are low, but not much lower at 80 °C than at 25 °C. This shows the partially protective effect of siderite deposits, which are much greater at 80 °C [35] and compensate for the higher corrosiveness of the fluid at this temperature.
- -
- The initiation of the steel corrosion with co-adsorption of hydrogen and sulfide during occurs the first hours of immersion of the steel. This results in a reduction and thus maintenance at a constant value of the corrosion rate of the steel.
- -
- The second stage involves the formation of iron sulfide deposits on the surface of the steel (see Table A2 and Table A5), which also slightly polarize the steel. In this case, we transition from a first type electrode to a second type electrode (Fe/FeS/S2−) for iron oxidation. The solution quickly darkens due to the formation of large quantities of iron sulfides (availability of Fe2+ due to polarization at 200 mV/OCP) and sulfides.
- -
- The last phase corresponds to the progressive crystallization of sulfide deposits from the FeS form to the FexSy form [16]. In presence of small amount of sulfides due to H2 production from corrosion and microbial activity, magnetite was observed in Cox water, while denser pyrite was found at 90 °C in relatively long term experiments (3 months) [16]. In this case, the pyrite formation at 90 °C inhibits the corrosion reaction. In our short-term and electrochemically aggressive experiment, with a CO2/H2S ratio of 25, mackinawite (FeS), which is weakly protective, is indeed the main corrosion product formed at 25 °C. The solubility of H2S is lower at 80 °C than at 25 °C, as highlighted in the PhreeqC simulations. However, FeS deposits, which are electroactive, become more reactive at higher temperatures, leading to an acceleration of corrosion at elevated temperatures. Furthermore, for pH values between 4 and 5.5, FeS protective films are unstable and partially dissolve, exposing carbon steel to a faster uniform attack [75]. The presence of iron sulfides is clearly evidenced. Unlike condition A, which does not contain sulfides, siderite (FeCO3) precipitation is unstable in the presence of sulfides. Even at 80 °C [75], where siderite normally forms in greater amounts, it remains a minor phase. At 80 °C, mackinawite transforms into more crystalline phases. However, due to their fissured and fragile nature, they promote localized corrosion, which can further accelerate the overall corrosion process. The progressive increase and transformation of amorphous FeS into crystalline, conductive mackinawite (pyrrhotite and pyrite in actual geothermal waters) leads to a continuous and severe attack on carbon steel [48].
- -
- The first stage involves the initiation of the steel corrosion. The O2 is available, and the corrosion rate is initially very high (hundreds of µA/cm2 at this stage).
- -
- The second stage involves the formation of corrosion deposits at the steel surface highlighted by the EIS in both cases (25 °C and 80 °C). This is correlated with a drop in corrosion rate measurements. Note that the appearance of the deposit using the EIS diagram at 80 °C occurred after 48 h of immersion, whereas it occurred in the first hours at 25 °C. With the PhreeqC simulations made (see Table A3 and Table A6), the corrosion products expected in these cases (large presence of oxygen) are ferrous oxides (magnetite and hematite) at 25 °C. At 80 °C, hematite is expected, but with a much lower SI (3.83 at 80 °C versus 14.7 at 25 °C), and no magnetite, consistent with the latter appearance of a new interface at the steel surface in the high temperature experiment (80 °C).
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temp. (°C) | [Fe2+] tot | [Fe3+] tot | [H2S] (M) | [HS−] (M) | [CO2] | [HCO3−] |
|---|---|---|---|---|---|---|
| 25 | 9.400 × 10−5 | 0 | 0 | 0 | 3.322 × 10−4 | 1.081 × 10−3 |
| 80 | 9.400 × 10−5 | 0 | 0 | 0 | 1.251 × 10−4 | 9.983 × 10−4 |
| Temp. (°C) | Phase Name | SI | log IAP | log K (298 or 353 K, 1 atm) | ||
| 25 | Calcite, CaCO3 | −0.62 | 1.22 | 1.85 | ||
| Siderite, FeCO3 | −0.76 | −1.03 | −0.27 | |||
| 80 | Calcite, CaCO3 | 0.40 | 1.45 | 1.05 | ||
| Siderite, FeCO3 | 0.23 | −0.91 | −1.14 | |||
| Temp. (°C) | [Fe2+] tot | [Fe3+] tot | [H2S] (M) | [HS−] (M) | [CO2] | [HCO3−] |
|---|---|---|---|---|---|---|
| 25 | 9.400 × 10−5 | 0 | 7.825 × 10−4 | 3.493 × 10−5 | 6.642 × 10−3 | 1.291 × 10−3 |
| 80 | 9.400 × 10−5 | 0 | 3.089 × 10−4 | 7.211 × 10−5 | 2.503 × 10−3 | 1.010 × 10−3 |
| Temp. (°C) | Phase Name | SI | log IAP | log K (298 or 353 K, 1 atm) | ||
| 25 | FeS(am), FeS | −0.76 | −3.7 | −2.99 | ||
| Mackinawite, FeS | −0.22 | −3.75 | −3.54 | |||
| Greisite, Fe3S4 | 0.98 | −20.91 | −21.89 | |||
| 80 | FeS(am), FeS | 0.12 | −3.33 | −3.45 | ||
| Mackinawite, FeS | 0.59 | −3.35 | −3.92 | |||
| Greisite, Fe3S4 | 1.46 | −19.83 | −21.29 | |||
| Temp. (°C) | [Fe2+] tot | [Fe3+] tot | [H2S] (M) | [HS−] (M) | [CO2] | [HCO3−] |
|---|---|---|---|---|---|---|
| 25 | 0 | 9.401 × 10−5 | 0 | 0 | 1.291 × 10−3 | 1.145 × 10−8 |
| 80 | 0 | 9.401 × 10−5 | 0 | 0 | 1.290 × 10−3 | 1.637 × 10−7 |
| Temp. (°C) | Phase Name | SI | log IAP | log K (298 or 353 K, 1atm) | ||
| 25 | Maghemite disord. (Fe2O3) | −7.64 | −4.80 | 2.84 | ||
| Hematite (Fe2O3) | 4.75 | −4.80 | −0.04 | |||
| Magnetite Fe3O4 | −25.15 | −14.79 | 10.36 | |||
| Magnetite(am) Fe3O4 | −29.38 | −14.79 | 14.59 | |||
| Goethite, FeOOH | −2.76 | −2.40 | 0.36 | |||
| Lepidocrocite, FeOOH | −4.25 | −2.40 | 1.85 | |||
| Calcite, CaCO3 | −11.15 | −9.30 | 1.85 | |||
| Siderite, FeCO3 | −18.94 | −19.21 | −0.27 | |||
| 80 | Maghemite disord. (Fe2O3) | 1.48 | 0.30 | −1.19 | ||
| Hematite (Fe2O3) | 3.87 | 0.30 | −3.58 | |||
| Magnetite Fe3O4 | −8.83 | −4.36 | 4.47 | |||
| Magnetite(am) Fe3O4 | −12.40 | −4.36 | 8.04 | |||
| Goethite, FeOOH | 1.43 | 0.15 | −1.29 | |||
| Lepidocrocite, FeOOH | 0.23 | 0.15 | −0.08 | |||
| Calcite, CaCO3 | −8.11 | −7.06 | 1.05 | |||
| Siderite, FeCO3 | −12.72 | −13.86 | −1.14 | |||
| Temp.(°C) | [Fe2+] tot | [Fe3+] tot | [H2S] (M) | [HS−] (M) | [CO2] | [HCO3−] |
|---|---|---|---|---|---|---|
| 25 | 9.319 × 10−4 | 8.077 × 10−6 | 0 | 0 | 3.321 × 10−4 | 2.512 × 10−3 |
| 80 | 7.691 × 10−4 | 1.709 × 10−4 | 0 | 0 | 1.251 × 10−4 | 1.888 × 10−3 |
| Temp.(°C) | Phase Name | SI | log IAP | log K (298 or 353 K, 1 atm) | ||
| 25 | Maghemite disord. (Fe2O3) | 11.82 | 14.66 | 2.84 | ||
| Hematite (Fe2O3) | 14.70 | 14.66 | −0.04 | |||
| Magnetite Fe3O4 | 14.80 | 25.16 | 10.36 | |||
| Magnetite(am) Fe3O4 | 10.57 | 25.16 | 14.59 | |||
| Goethite, FeOOH | 6.97 | 7.33 | 0.36 | |||
| Lepidocrocite, FeOOH | 5.48 | 7.33 | 1.85 | |||
| Calcite, CaCO3 | 0.11 | 1.95 | 1.85 | |||
| Siderite, FeCO3 | 0.95 | 0.68 | −0.27 | |||
| 80 | Maghemite disord. (Fe2O3) | 13.48 | 12.29 | −1.19 | ||
| Hematite, Fe2O3 | 7.43 | 6.15 | −1.29 | |||
| Magnetite, Fe3O4 | 18.57 | 23.04 | 4.47 | |||
| Magnetite(am), Fe3O4 | 14.99 | 23.04 | 8.04 | |||
| Goethite, FeOOH | 15.87 | 12.29 | −3.58 | |||
| Lepidocrocite, FeOOH | 6.23 | 6.15 | −0.08 | |||
| Calcite, CaCO3 | 0.95 | 2.00 | 1.05 | |||
| Siderite, FeCO3 | 1.66 | 0.52 | −1.14 | |||
| Temp.(°C) | [Fe2+] tot | [Fe3+] tot | [H2S] (M) | [HS−] (M) | [CO2] | [HCO3−] |
|---|---|---|---|---|---|---|
| 25 | 9.400 × 10−4 | 0 | 7.825 × 10−4 | 7.387 × 10−5 | 6.640 × 10−3 | 2.729 × 10−3 |
| 80 | 9.400 × 10−4 | 0 | 3.089 × 10−4 | 1.668 × 10−4 | 2.502 × 10−3 | 2.335 × 10−3 |
| Temp.(°C) | Phase Name | SI | log IAP | log K (298 or 353 K, 1 atm) | ||
| 25 | FeS(am), FeS | 0.88 | −2.11 | −2.99 | ||
| Mackinawite, FeS | 1.43 | −2.11 | −3.54 | |||
| Greisite, Fe3S4 | 5.76 | −16.13 | −21.89 | |||
| Calcite, CaCO | −1.13 | 0.72 | 1.85 | |||
| Siderite, FeCO3 | −0.26 | −0.53 | −0.27 | |||
| 80 | FeS(am), FeS | 1.85 | −1.61 | −3.45 | ||
| Mackinawite, FeS | 2.31 | −1.61 | −3.92 | |||
| Greisite, Fe3S4 | 6.45 | −14.85 | −21.29 | |||
| Calcite, CaCO3 | −0.16 | 0.88 | 1.05 | |||
| Siderite, FeCO3 | 0.69 | −0.45 | −1.14 | |||
| Temp.(°C) | [Fe2+] tot | [Fe3+] tot | [H2S] (M) | [HS−] (M) | [CO2] | [HCO3−] |
|---|---|---|---|---|---|---|
| 25 | 0 | 9.409 × 10−4 | 0 | 0 | 1.291 × 10−3 | 1.156 × 10−8 |
| 80 | 0 | 9.401 × 10−4 | 0 | 0 | 1.290 × 10−3 | 1.738 × 10−7 |
| Temp.(°C) | Phase Name | SI | log IAP | log K (298 or 353 K, 1 atm) | ||
| 25 | Maghemite disord. (Fe2O3) | −5.60 | −2.76 | 2.84 | ||
| Hematite (Fe2O3) | 2.72 | −2.76 | −0.04 | |||
| Magnetite Fe3O4 | −22.09 | −11.73 | 10.36 | |||
| Magnetite(am) Fe3O4 | −26.32 | −11.73 | 14.59 | |||
| Goethite, FeOOH | −1.74 | −1.38 | 0.36 | |||
| Lepidocrocite, FeOOH | −3.23 | −1.38 | 1.85 | |||
| Calcite, CaCO3 | −11.14 | −9.30 | 1.85 | |||
| Siderite, FeCO3 | −17.92 | −18.19 | −0.27 | |||
| 80 | Maghemite disord. (Fe2O3) | 3.63 | 2.44 | −1.19 | ||
| Hematite (Fe2O3) | 6.02 | 2.44 | −3.58 | |||
| Magnetite Fe3O4 | −5.61 | −1.14 | 4.47 | |||
| Magnetite(am) Fe3O4 | −9.18 | −1.14 | 8.04 | |||
| Goethite, FeOOH | 2.51 | 1.22 | −1.29 | |||
| Lepidocrocite, FeOOH | 1.30 | 1.22 | −0.08 | |||
| Calcite, CaCO3 | −8.06 | −7.01 | 1.05 | |||
| Siderite, FeCO3 | −11.65 | −12.78 | −1.14 | |||

References
- Altmann, S. ’Geo’chemical Research: A Key Building Block for Nuclear Waste Disposal Safety Cases. J. Contam. Hydrol. 2008, 102, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Shi, J.; Chen, Y.; Li, Z. Effect of Chemical Corrosion on the Mechanical Characteristics of Parent Rocks for Nuclear Waste Storage. Sci. Technol. Nucl. Install. 2016, 2016, 7853787. [Google Scholar] [CrossRef]
- Jonsson, M. Radiation Effects on Materials Used in Geological Repositories for Spent Nuclear Fuel. Int. Sch. Res. Not. 2012, 2012, 639520. [Google Scholar] [CrossRef]
- Gaucher, E.; Robelin, C.; Matray, J.M.; Négrel, G.; Gros, Y.; Heitz, J.F.; Vinsot, A.; Rebours, H.; Cassagnabère, A.; Bouchet, A. ANDRA Underground Research Laboratory: Interpretation of the Mineralogical and Geochemical Data Acquired in the Callovian–Oxfordian Formation by Investigative Drilling. Phys. Chem. Earth Parts A/B/C 2004, 29, 55–77. [Google Scholar] [CrossRef]
- Gaucher, É.C.; Blanc, P.; Bardot, F.; Braibant, G.; Buschaert, S.; Crouzet, C.; Gautier, A.; Girard, J.-P.; Jacquot, E.; Lassin, A.; et al. Modelling the porewater chemistry of the Callovian–Oxfordian formation at a regional scale. Comptes Rendus. Géoscience 2006, 338, 917–930. [Google Scholar] [CrossRef]
- Gaucher, E.C.; Tournassat, C.; Pearson, F.J.; Blanc, P.; Crouzet, C.; Lerouge, C.; Altmann, S. A Robust Model for Pore-Water Chemistry of Clayrock. Geochim. Cosmochim. Acta 2009, 73, 6470–6487. [Google Scholar] [CrossRef]
- Fernández, A.M.; Sánchez-Ledesma, D.M.; Tournassat, C.; Melón, A.; Gaucher, E.C.; Astudillo, J.; Vinsot, A. Applying the Squeezing Technique to Highly Consolidated Clayrocks for Pore Water Characterisation: Lessons Learned from Experiments at the Mont Terri Rock Laboratory. Appl. Geochem. 2014, 49, 2–21. [Google Scholar] [CrossRef]
- Tournassat, C.; Bourg, I.C.; Steefel, C.I.; Bergaya, F. Chapter 1—Surface Properties of Clay Minerals. In Developments in Clay Science; Tournassat, C., Steefel, C.I., Bourg, I.C., Bergaya, F., Eds.; Natural and Engineered Clay Barriers; Elsevier: Amsterdam, The Netherlands, 2015; Volume 6, pp. 5–31. [Google Scholar]
- Vinsot, A.; Appelo, C.A.J.; Cailteau, C.; Wechner, S.; Pironon, J.; De Donato, P.; De Cannière, P.; Mettler, S.; Wersin, P.; Gäbler, H.-E. CO2 Data on Gas and Pore Water Sampled in Situ in the Opalinus Clay at the Mont Terri Rock Laboratory. Phys. Chem. Earth Parts A/B/C 2008, 33, S54–S60. [Google Scholar] [CrossRef]
- Vinsot, A.; Mettler, S.; Wechner, S. In Situ Characterization of the Callovo-Oxfordian Pore Water Composition. Phys. Chem. Earth Parts A/B/C 2008, 33, S75–S86. [Google Scholar] [CrossRef]
- Martin, F.A.; Perrin, S.; Bataillon, C. Evaluating the Corrosion Rate of Low Alloyed Steel in Callovo-Oxfordian Clay: Towards a Complementary EIS, Gravimetric and Structural Study. MRS Online Proc. Libr. 2012, 1475, 471–476. [Google Scholar] [CrossRef]
- Schlegel, M.L.; Necib, S.; Daumas, S.; Blanc, C.; Foy, E.; Trcera, N.; Romaine, A. Microstructural Characterization of Carbon Steel Corrosion in Clay Borehole Water under Anoxic and Transient Acidic Conditions. Corros. Sci. 2016, 109, 126–144. [Google Scholar] [CrossRef]
- Necib, S.; Linard, Y.; Crusset, D.; Schlegel, M.; Daumas, S.; Michau, N. Corrosion Processes of C-Steel in Long-Term Repository Conditions. Corros. Eng. Sci. Technol. 2017, 52, 127–130. [Google Scholar] [CrossRef]
- Necib, S.; Linard, Y.; Crusset, D.; Michau, N.; Daumas, S.; Burger, E.; Romaine, A.; Schlegel, M.L. Corrosion at the Carbon Steel–clay Borehole Water and Gas Interfaces at 85 °C under Anoxic and Transient Acidic Conditions. Corros. Sci. 2016, 111, 242–258. [Google Scholar] [CrossRef]
- Hajj, H.E.; Abdelouas, A.; Grambow, B.; Martin, C.; Dion, M. Microbial Corrosion of P235GH Steel under Geological Conditions. Phys. Chem. Earth Parts A/B/C 2010, 35, 248–253. [Google Scholar] [CrossRef]
- Mendili, Y.E.; Abdelouas, A. Corrosion of P235GH Carbon Steel in Simulated Bure Soil Solution. J. Mater. Environ. Sci 2013, 4, 786–791. [Google Scholar]
- El Mendili, Y.; Abdelouas, A.; Karakurt, G.; Aït Chaou, A.; Essehli, R.; Bardeau, J.-F.; Grenèche, J.-M. The Effect of Temperature on Carbon Steel Corrosion under Geological Conditions. Appl. Geochem. 2015, 52, 76–85. [Google Scholar] [CrossRef]
- Sano Moyeme, Y.C.; Betelu, S.; Bertrand, J.; Groenen Serrano, K.; Ignatiadis, I. Corrosion Current Density of API 5L X65 Carbon Steel in Contact with Natural Callovian-Oxfordian Clay Pore Water, Assessed by Various Electrochemical Methods over 180 Days. Metals 2023, 13, 966. [Google Scholar] [CrossRef]
- Lotz, H.; Neff, D.; Mercier-Bion, F.; Bataillon, C.; Dillmann, P.; Gardés, E.; Monnet, I.; Dynes, J.J.; Foy, E. Localised Corrosion of Iron and Steel in the Callovo-Oxfordian Porewater after 3 Months at 120 C: Characterizations at Micro and Nanoscale and Formation Mechanisms. Corros. Sci. 2023, 219, 111235. [Google Scholar] [CrossRef]
- Comeaux, R.V. The Role of Oxygen in Corrosion and Cathodic Protection. Corrosion 1952, 8, 305–310. [Google Scholar] [CrossRef]
- Ruther, W.E.; Hart, R.K. Influence of Oxygen on High Temperature Aqueous Corrosion of Iron. Corrosion 1963, 19, UAC-6956. [Google Scholar] [CrossRef]
- Strauss, M.B.; Bloom, M.C. Corrosion Mechanisms in the Reaction of Steel with Water and Oxygenated Solutions at Room Temperature and 316 °C. J. Electrochem. Soc. 1960, 107, 73. [Google Scholar] [CrossRef]
- Cox, G.L.; Roetheli, B.E. Effect of Oxygen Concentration on Corrosion Rates of Steel and Composition of Corrosion Products Formed in Oxygenated Water. Ind. Eng. Chem. 1931, 23, 1012–1016. [Google Scholar] [CrossRef]
- Tromans, D. Modeling Oxygen-Controlled Corrosion of Steels in Hot Waters. Corrosion 1999, 55, 942–947. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhang, T.C. Effects of Dissolved Oxygen on Formation of Corrosion Products and Concomitant Oxygen and Nitrate Reduction in Zero-Valent Iron Systems with or without Aqueous Fe2+. Water Res. 2005, 39, 1751–1760. [Google Scholar] [CrossRef]
- El Hajj, H.; Abdelouas, A.; El Mendili, Y.; Karakurt, G.; Grambow, B.; Martin, C. Corrosion of Carbon Steel under Sequential Aerobic–Anaerobic Environmental Conditions. Corros. Sci. 2013, 76, 432–440. [Google Scholar] [CrossRef]
- Saha, J.K. Corrosion of Constructional Steels in Marine and Industrial Environment: Frontier Work in Atmospheric Corrosion; Engineering Materials; Springer: Chennai, India, 2013; ISBN 978-81-322-0719-1. [Google Scholar]
- Lu, W.-F.; Chen, T.-C.; Tsai, K.-C.; Yung, T.-Y. The Corrosion Behavior of Carbon Steel Materials Used at Nuclear Power Plants During Deactivation and Decommissioning Processes. Metals 2024, 14, 1444. [Google Scholar] [CrossRef]
- Tran, T.; Brown, B.; Nesic, S. Corrosion of Mild Steel in an Aqueous CO2 Environment–Basic Electrochemical Mechanisms Revisited. In Corrosion Conference and Expo 2015, Proceedings of the Nace Corrosion, Dallas, TX, USA, 5–19 March 2015; NACE: Houston, TX, USA, 2015; p. NACE-2015. [Google Scholar]
- Basilico, E.; Marcelin, S.; Mingant, R.; Kittel, J.; Fregonese, M.; Ropital, F. The Effect of Chemical Species on the Electrochemical Reactions and Corrosion Product Layer of Carbon Steel in CO2 Aqueous Environment: A Review. Mater. Corros. 2021, 72, 1152–1167. [Google Scholar] [CrossRef]
- Shayegani, M.; Afshar, A.; Ghorbani, M.; Rahmaniyan, M. Mild Steel Carbon Dioxide Corrosion Modelling in Aqueous Solutions. Corros. Eng. Sci. Technol. 2008, 43, 290–296. [Google Scholar] [CrossRef]
- Ogundele, G.I.; White, W.E. Some Observations on Corrosion of Carbon Steel in Aqueous Environments Containing Carbon Dioxide. Corrosion 1986, 42, 71–78. [Google Scholar] [CrossRef]
- Yang, S.; Li, M.; Liu, T.; Gao, P.; Zhang, H.; Yin, C.; Tang, Y. Protective Mechanism of Carbon Dioxide in Pipelines for Water Containing Typical Corrosive Anions. Process Saf. Environ. Prot. 2024, 188, 654–663. [Google Scholar] [CrossRef]
- Refait, P.; Grolleau, A.-M.; Jeannin, M.; Rémazeilles, C.; Sabot, R. Corrosion of Carbon Steel in Marine Environments: Role of the Corrosion Product Layer. Corros. Mater. Degrad. 2020, 1, 198–218. [Google Scholar] [CrossRef]
- Taleb, W.; Pessu, F.; Wang, C.; Charpentier, T.; Barker, R.; Neville, A. Siderite Micro-Modification for Enhanced Corrosion Protection. npj Mater. Degrad. 2017, 1, 1–6. [Google Scholar] [CrossRef]
- da Silva, P.N.; Dugstad, A.; Svenningsen, G.; da Cunha Ponciano Gomes, J.A. The Influence of Oxygen on Protective Iron Carbonate Scales Formed on Carbon Steel in CO2 Environments at Near-Neutral pH. In Proceedings of the AMPP Annual Conference + Expo, San Antonio, TX, USA, 6 March 2022. [Google Scholar]
- Ma, H.; Cheng, X.; Li, G.; Chen, S.; Quan, Z.; Zhao, S.; Niu, L. The Influence of Hydrogen Sulfide on Corrosion of Iron under Different Conditions. Corros. Sci. 2000, 42, 1669–1683. [Google Scholar] [CrossRef]
- Gao, S.; Brown, B.; Young, D.; Nesic, S.; Singer, M. Formation Mechanisms of Iron Oxide and Iron Sulfide at High Temperature in Aqueous H2S Corrosion Environment. J. Electrochem. Soc. 2018, 165, C171. [Google Scholar] [CrossRef]
- Cheng, X.L.; Ma, H.Y.; Zhang, J.P.; Chen, X.; Chen, S.H.; Yang, H.Q. Corrosion of Iron in Acid Solutions with Hydrogen Sulfide. Corrosion 1998, 54, 369–376. [Google Scholar] [CrossRef]
- Yoo, J.-S.; Chung, N.T.; Lee, Y.-H.; Kim, Y.-W.; Kim, J.-G. Effect of Sulfide and Chloride Ions on Pitting Corrosion of Type 316 Austenitic Stainless Steel in Groundwater Conditions Using Response Surface Methodology. Materials 2024, 17, 178. [Google Scholar] [CrossRef]
- Ma, H.; Cheng, X.; Chen, S.; Wang, C.; Zhang, J.; Yang, H. An Ac Impedance Study of the Anodic Dissolution of Iron in Sulfuric Acid Solutions Containing Hydrogen Sulfide. J. Electroanal. Chem. 1998, 451, 11–17. [Google Scholar] [CrossRef]
- Romaine, A. Role of Sulfide Species in the Corrosion of Non-Alloy Steel: Heterogeneities of the Layer of Corrosion Products and Galvanic Coupling. Ph.D. Thesis, Universiy of La Rochelle, La Rochelle, France, 2014. [Google Scholar]
- Somervuori, M.; Isotahdon, E.; Nuppunen-Puputti, M.; Bomberg, M.; Carpén, L.; Rajala, P. A Comparison of Different Natural Groundwaters from Repository Sites—Corrosivity, Chemistry and Microbial Community. Corros. Mater. Degrad. 2021, 2, 603–624. [Google Scholar] [CrossRef]
- Konovalova, V. The Effect of Temperature on the Corrosion Rate of Iron-Carbon Alloys. Mater. Today: Proc. 2021, 38, 1326–1329. [Google Scholar] [CrossRef]
- Betelu, S.; Tournassat, C.; Bertrand, J.; Ignatiadis, I. Pyrite-Electrode Potential Measurements in Reference Solutions versus Measured and Computed Redox Potential: At the Roots of an Innovative Generic pH Electrode for Nuclear Waste Repositories. In Proceedings of the 12th International High-Level Radioactive Waste Management Conference, Albuquerque, NM, USA, 29 April–3 May 2012. [Google Scholar]
- Blanc, P.; Lassin, A.; Piantone, P.; Azaroual, M.; Jacquemet, N.; Fabbri, A.; Gaucher, E.C. Thermoddem: A Geochemical Database Focused on Low Temperature Water/Rock Interactions and Waste Materials. Appl. Geochem. 2012, 27, 2107–2116. [Google Scholar] [CrossRef]
- Castillo, C.; Ignatiadis, I. Sulfate-Reduction State of the Geothermal Dogger Aquifer, Paris Basin, France After 35 Years of Exploitation: Analysis And Consequences of Bacterial Proliferation in Casings and Reservoir. In Proceedings of the Thirty-Seventh Workshop on Geothermal Reservoir Engineering, Stanford, CA, USA, 30 January–1 February 2012. [Google Scholar]
- Betelu, S.; Helali, C.; Ignatiadis, I. Laboratory-Scale Implementation of Standardized Reconstituted Geothermal Water for Electrochemical Investigations of Carbon Steel Corrosion. Metals 2024, 14, 1216. [Google Scholar] [CrossRef]
- Gabrielli, C.; Takenouti, H. Méthodes Électrochimiques Appliquées à La Corrosion-Techniques Dynamiques. Les Tech. L’ingenieur 2010, COR 811, 1–22. [Google Scholar] [CrossRef]
- Reeve, J.C.; Bech-Nielsen, G. The Stern-Geary Method. Basic Difficulties and Limitations and a Simple Extension Providing Improved Reliability. Corros. Sci. 1973, 13, 351–359. [Google Scholar] [CrossRef]
- Angst, U.; Büchler, M. On the Applicability of the Stern–Geary Relationship to Determine Instantaneous Corrosion Rates in Macro-Cell Corrosion. Mater. Corros. 2015, 66, 1017–1028. [Google Scholar] [CrossRef]
- Mansfeld, F.; Oldham, K.B. A Modification of the Stern—Geary Linear Polarization Equation. Corros. Sci. 1971, 11, 787–796. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications. Surf. Technol. 1983, 20, 91–92. [Google Scholar]
- Panchenko, Y.M.; Marshakov, A.I.; Igonin, T.N.; Kovtanyuk, V.V.; Nikolaeva, L.A. Long-Term Forecast of Corrosion Mass Losses of Technically Important Metals in Various World Regions Using a Power Function. Corros. Sci. 2014, 88, 306–316. [Google Scholar] [CrossRef]
- Jiao, D.; Rempe, S.B. CO2 Solvation Free Energy Using Quasi-Chemical Theory. J. Chem. Phys. 2011, 134, 224506. [Google Scholar] [CrossRef]
- Duan, Z.; Sun, R.; Liu, R.; Zhu, C. Accurate Thermodynamic Model for the Calculation of H2S Solubility in Pure Water and Brines. Energy Fuels 2007, 21, 2056–2065. [Google Scholar] [CrossRef]
- Daoudi, J.; Betelu, S.; Tzedakis, T.; Bertrand, J.; Ignatiadis, I. A Multi-Parametric Device with Innovative Solid Electrodes for Long-Term Monitoring of pH, Redox-Potential and Conductivity in a Nuclear Waste Repository. Sensors 2017, 17, 1372. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.E. Boyd, C.E., Ed.; Dissolved Oxygen and Redox Potential. In Water Quality: An Introduction; Springer: Boston, MA, USA, 2000; pp. 69–94. ISBN 978-1-4615-4485-2. [Google Scholar]
- Aslam, R. Chapter 2—Potentiodynamic Polarization Methods for Corrosion Measurement. In Electrochemical and Analytical Techniques for Sustainable Corrosion Monitoring; Aslam, J., Verma, C., Mustansar Hussain, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 25–37. ISBN 978-0-443-15783-7. [Google Scholar]
- Zhang, X.L.; Jiang, Z.H.; Yao, Z.P.; Song, Y.; Wu, Z.D. Effects of Scan Rate on the Potentiodynamic Polarization Curve Obtained to Determine the Tafel Slopes and Corrosion Current Density. Corros. Sci. 2009, 51, 581–587. [Google Scholar] [CrossRef]
- Zhou, H.; Chhin, D.; Morel, A.; Gallant, D.; Mauzeroll, J. Potentiodynamic Polarization Curves of AA7075 at High Scan Rates Interpreted Using the High Field Model. npj Mater. Degrad. 2022, 6, 1–11. [Google Scholar] [CrossRef]
- Frankel, G.S. Hughes, A.E., Mol, J.M.C., Zheludkevich, M.L., Buchheit, R.G., Eds.; Fundamentals of Corrosion Kinetics. In Active Protective Coatings; Springer Series in Materials Science; Springer: Dordrecht, The Netherlands, 2016; Volume 233, pp. 17–32. ISBN 978-94-017-7538-0. [Google Scholar]
- Poorqasemi, E.; Abootalebi, O.; Peikari, M.; Haqdar, F. Investigating Accuracy of the Tafel Extrapolation Method in HCl Solutions. Corros. Sci. 2009, 51, 1043–1054. [Google Scholar] [CrossRef]
- De Oliveira, M.C.; Figueredo, R.M.; Acciari, H.A.; Codaro, E.N. Corrosion Behavior of API 5L X65 Steel Subject to Plastic Deformation. J. Mater. Res. Technol. 2018, 7, 314–318. [Google Scholar] [CrossRef]
- Zhang, R.; Sur, D.; Li, K.; Witt, J.; Black, R.; Whittingham, A.; Scully, J.R.; Hattrick-Simpers, J. Bayesian Assessment of Commonly Used Equivalent Circuit Models for Corrosion Analysis in Electrochemical Impedance Spectroscopy. npj Mater. Degrad. 2024, 8, 1–7. [Google Scholar] [CrossRef]
- McCafferty, E.; Hackerman, N. Double Layer Capacitance of Iron and Corrosion Inhibition with Polymethylene Diamines. J. Electrochem. Soc. 1972, 119, 146. [Google Scholar] [CrossRef]
- Bonnel, A.; Dabosi, F.; Deslouis, C.; Duprat, M.; Keddam, M.; Tribollet, B. Corrosion Study of a Carbon Steel in Neutral Chloride Solutions by Impedance Techniques. J. Electrochem. Soc. 1983, 130, 753. [Google Scholar] [CrossRef]
- Guo, X.-P.; Tomoe, Y. The Effect of Corrosion Product Layers on the Anodic and Cathodic Reactions of Carbon Steel in CO2-Saturated Mdea Solutions at 100 °C. Corros. Sci. 1999, 41, 1391–1402. [Google Scholar] [CrossRef]
- Flis, J.; Dawson, J.L.; Gill, J.; Wood, G.C. Impedance and Electrochemical Noise Measurements on Iron and Iron-Carbon Alloys in Hot Caustic Soda. Corros. Sci. 1991, 32, 877–892. [Google Scholar] [CrossRef]
- Christensen, B.J.; Mason, T.O.; Jennings, H.M. Influence of Silica Fume on the Early Hydration of Portland Cements Using Impedance Spectroscopy. J. Am. Ceram. Soc. 1992, 75, 939–945. [Google Scholar] [CrossRef]
- Ma, H.Y.; Cheng, X.L.; Chen, S.H.; Li, G.Q.; Chen, X.; Lei, S.B.; Yang, H.Q. Theoretical Interpretation on Impedance Spectra for Anodic Iron Dissolution in Acidic Solutions Containing Hydrogen Sulfide. Corrosion 1998, 54, 634–640. [Google Scholar] [CrossRef]
- Mansfeld, F. Discussion: Electrochemical Techniques for Studying Corrosion of Reinforcing Steel: Limitations and Advantages. Corrosion 2005, 61, 739–742. [Google Scholar] [CrossRef]
- Epelboin, I.; Gabrielli, C.; Keddam, M.; Takenouti, H. The Study of the Passivation Process by the Electrode Impedance Analysis. In Electrochemical Materials Science; Bockris, J.O., Conway, B.E., Yeager, E., White, R.E., Eds.; Springer: Boston, MA, USA, 1981; pp. 151–192. ISBN 978-1-4757-4827-7. [Google Scholar]
- El-Feki, A.A.; Walter, G.W. Corrosion Rate Measurements under Conditions of Mixed Charge Transfer plus Diffusion Control Including the Cathodic Metal Ion Deposition Partial Reaction. Corros. Sci. 2000, 42, 1055–1070. [Google Scholar] [CrossRef]
- Choi, Y.-S.; Hassani, S.; Vu, T.N.; Nešić, S.B.; Abas, A.Z.; Nor, A.M.; Suhor, M.F. Strategies for Corrosion Inhibition of Carbon Steel Pipelines Under Supercritical CO2/H2S Environments. Corrosion 2019, 75, 1156–1172. [Google Scholar] [CrossRef]










| Elements or Chemistry | Concentration (mM) or Value | Elements | Concentration (mM) |
|---|---|---|---|
| pH | 7.35 | K | 7.07 |
| Redox potential (mV/SHE) | −180 | Ca | 14.8 |
| Ionic strength | 116.0 | Mg | 14.1 |
| C(4) or carbonate | 1.29 | Sr | 1.12 |
| S(6) or sulfate | 34.0 | Si | 0.0943 |
| Cl− | 30.1 | Fe | 0.0940 |
| Na | 32.0 | Al | 0.0000086 |
| Mineral Salts Names and Formulas | Mass (g) for 1 L of Deionized Water |
|---|---|
| Sodium bicarbonate, NaHCO3 | 0.445 |
| Potassium chloride, KCl | 0.186 |
| Magnesium chloride hydrate MgCl2, 6H2O | 1.830 |
| Ammonium sulfate (NH4+)2, SO42− | 0.073 |
| Calcium sulfate CaSO4,2H2O | 1.416 |
| Calcium chloride, CaCl2 (anhydrous) | 1.887 |
| Sodium chloride, NaCl | 14.164 |
| Elements | Chemical Composition (% Mass) |
|---|---|
| C | 0.16 |
| Mn | 1.65 |
| Si | 0.45 |
| Ti | 0.06 |
| P | 0.02 |
| S | 0.01 |
| V | 0.07 |
| Nb | 0.05 |
| Fe | ~97.53 |
| Circuit Element | Immersion Time | |||
|---|---|---|---|---|
| 24 h | 96 h | |||
| Value | Relative Error (%) | Value | Relative Error (%) | |
| Rs (ohm) | 10.5 | 0.9 | 5.3 | 1.1 |
| Q-Yo | 6.407 10−3 | 1.4 | 2.042 10−2 | 1.4 |
| n | 0.81 | 0.7 | 0.774 | 0.8 |
| Rct (ohm) | 636 | 2.6 | 459 | 2.5 |
| L(H) | 1.029 × 104 | 17.1 | - | - |
| Rl (ohm) | 258 | 10.8 | - | - |
| Cdl calculated (F) | 4.35 10−3 | - | 6.9 10−3 | - |
| Circuit Element | Value | Error (%) |
|---|---|---|
| Rs (ohm) | 9.3 | 0.96 |
| Q-Yo | 2.701 × 10−3 | 4 |
| n | 0.99 | 0.9 |
| Rct (ohm) | 150.1 | 21 |
| Q-Yoi | 1.834 × 10−3 | 9.5 |
| ni | 0.94 | 5.2 |
| Ri (ohm) | 459.6 | 7.4 |
| Circuit Element | Value | Relative Error (%) |
|---|---|---|
| Rs (ohms) | 2.4 | 19.4 |
| Yo | 1.23 10−5 | 25.4 |
| nf | 0.84 | 3.7 |
| Rf (ohms) | 11 | 4.8 |
| Ydl | 1.83 10−3 | 1.5 |
| n | 0.79 | 0.6 |
| Rct (ohms) | 2155 | 0.9 |
| O-Yo | 0.11 | 1.1 |
| O-B | 11.6 | 4.2 |
| Condition | Corrosion Current Densities (µA cm−2) at 60 h | ||
|---|---|---|---|
| LPR | EIS | Tafel | |
| A-CO2 1%, N2 99%; 25 °C | 28 | 25 | 10 |
| A-CO2 1%, N2 99%; 80 °C | 11 | 32 | 7 |
| B- H2S 0.8%, CO2 20%, N2 79.2%; 25 °C | 43 | 39 | 37 |
| B- H2S 0.8%, CO2 20%, N2 79.2%; 80 °C | 138 | 132 | 103 |
| C-O2 20%, N2 80%; 25 °C | 62 | 81 | 37 |
| C-O2 20%, N2 80%; 80 °C | 236 | 580 | 435 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sano Moyeme, Y.C.; Betelu, S.; Bertrand, J.; Serrano, K.G.; Ignatiadis, I. Corrosion Behavior of X65 API 5L Carbon Steel Under Simulated Storage Conditions: Influence of Gas Mixtures, Redox States, and Temperature Assessed Using Electrochemical Methods for up to 100 Hours. Metals 2025, 15, 221. https://doi.org/10.3390/met15020221
Sano Moyeme YC, Betelu S, Bertrand J, Serrano KG, Ignatiadis I. Corrosion Behavior of X65 API 5L Carbon Steel Under Simulated Storage Conditions: Influence of Gas Mixtures, Redox States, and Temperature Assessed Using Electrochemical Methods for up to 100 Hours. Metals. 2025; 15(2):221. https://doi.org/10.3390/met15020221
Chicago/Turabian StyleSano Moyeme, Yendoube Charles, Stephanie Betelu, Johan Bertrand, Karine Groenen Serrano, and Ioannis Ignatiadis. 2025. "Corrosion Behavior of X65 API 5L Carbon Steel Under Simulated Storage Conditions: Influence of Gas Mixtures, Redox States, and Temperature Assessed Using Electrochemical Methods for up to 100 Hours" Metals 15, no. 2: 221. https://doi.org/10.3390/met15020221
APA StyleSano Moyeme, Y. C., Betelu, S., Bertrand, J., Serrano, K. G., & Ignatiadis, I. (2025). Corrosion Behavior of X65 API 5L Carbon Steel Under Simulated Storage Conditions: Influence of Gas Mixtures, Redox States, and Temperature Assessed Using Electrochemical Methods for up to 100 Hours. Metals, 15(2), 221. https://doi.org/10.3390/met15020221