
Study on the Microscopic Mechanism of the Grain Refinement of Al-Ti-B Master Alloy

Abstract
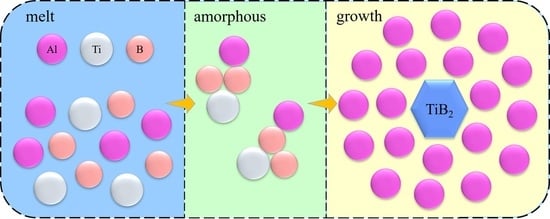
1. Introduction
2. Computational Details
2.1. Calculation of Clusters
2.2. Ab Initio Molecular Dynamics Calculation (AIMD)
3. Results and Discussion
3.1. The Ground State Structures of TinBn (n = 2–12) Clusters
3.2. Stability of TinBn (n = 2–12) Clusters
3.3. Prediction of Reaction Sites in TinBn (n = 2–12) Clusters
3.4. Ab Initio Molecular Dynamics Simulation Results
4. Conclusions
- (1)
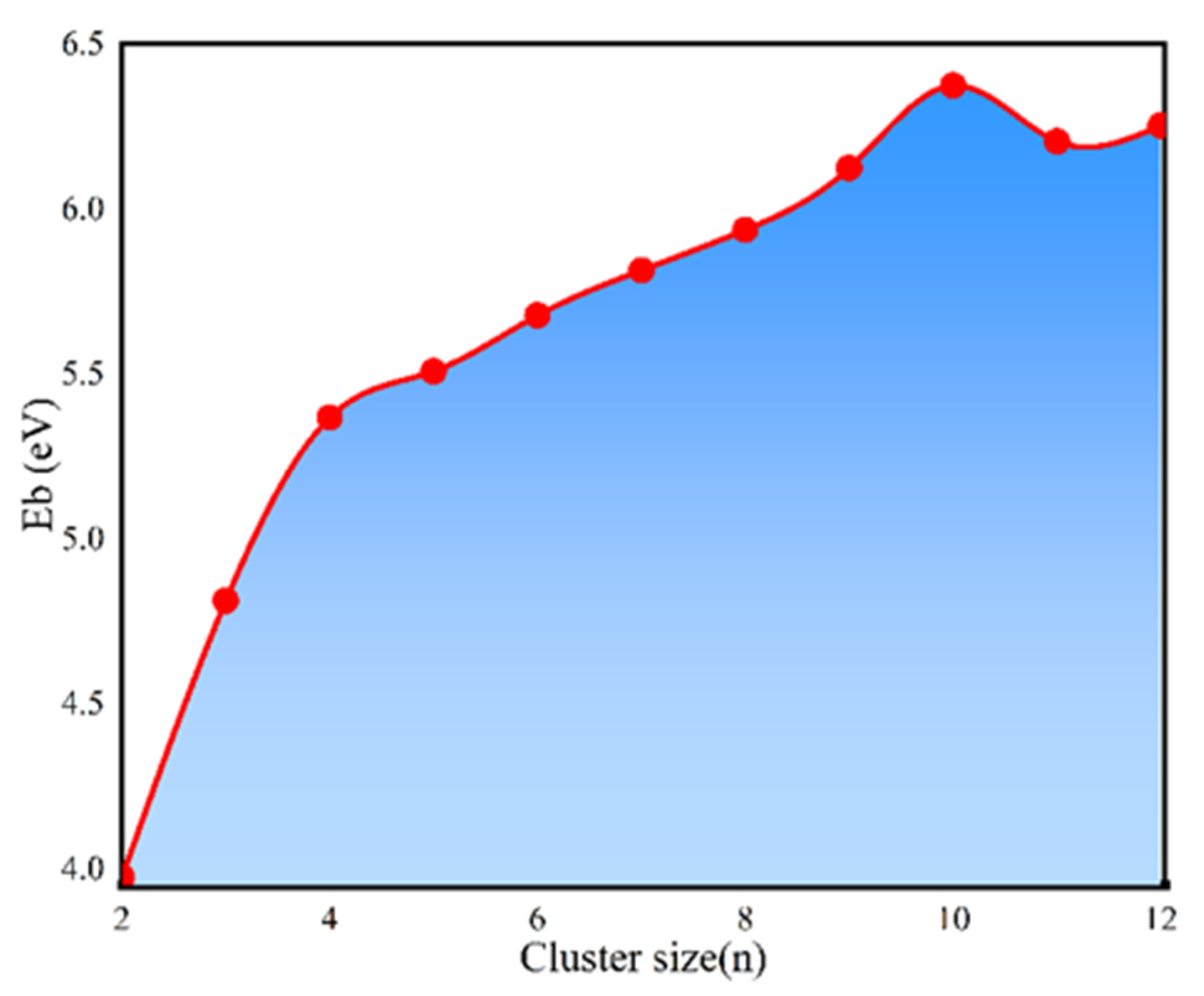
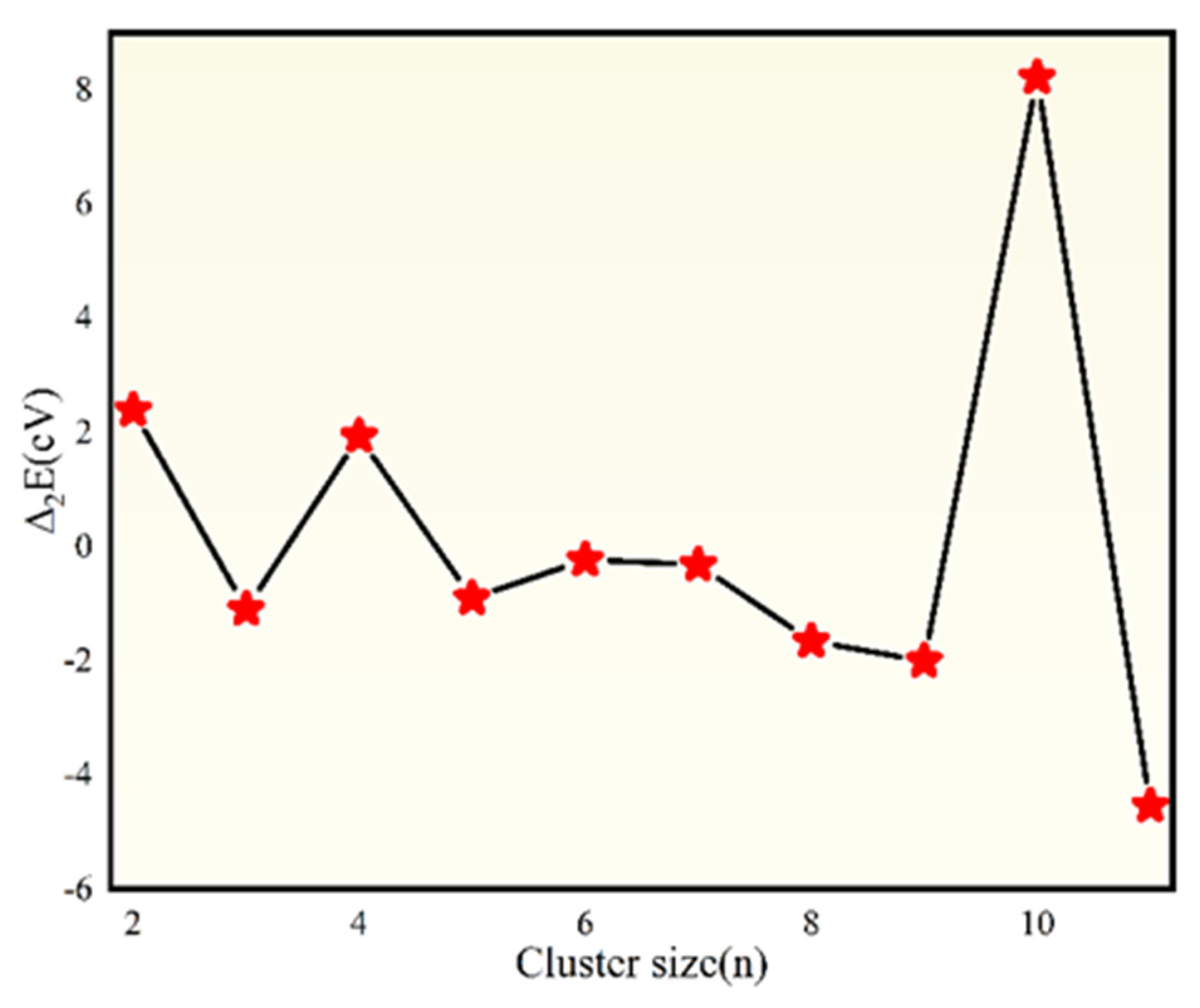
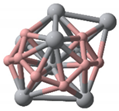
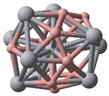
- TiB2 triangular structures are present in the cluster structures from n equal to 2 to 12; the amount of TiB2 increases as the cluster size increases and Ti atoms are oriented towards the surface of clusters. The average binding energy and second-order difference energy of the clusters show that the stability of the clusters increases with the increasing number of atoms, and the most stable structure is obtained at n = 10.
- (2)
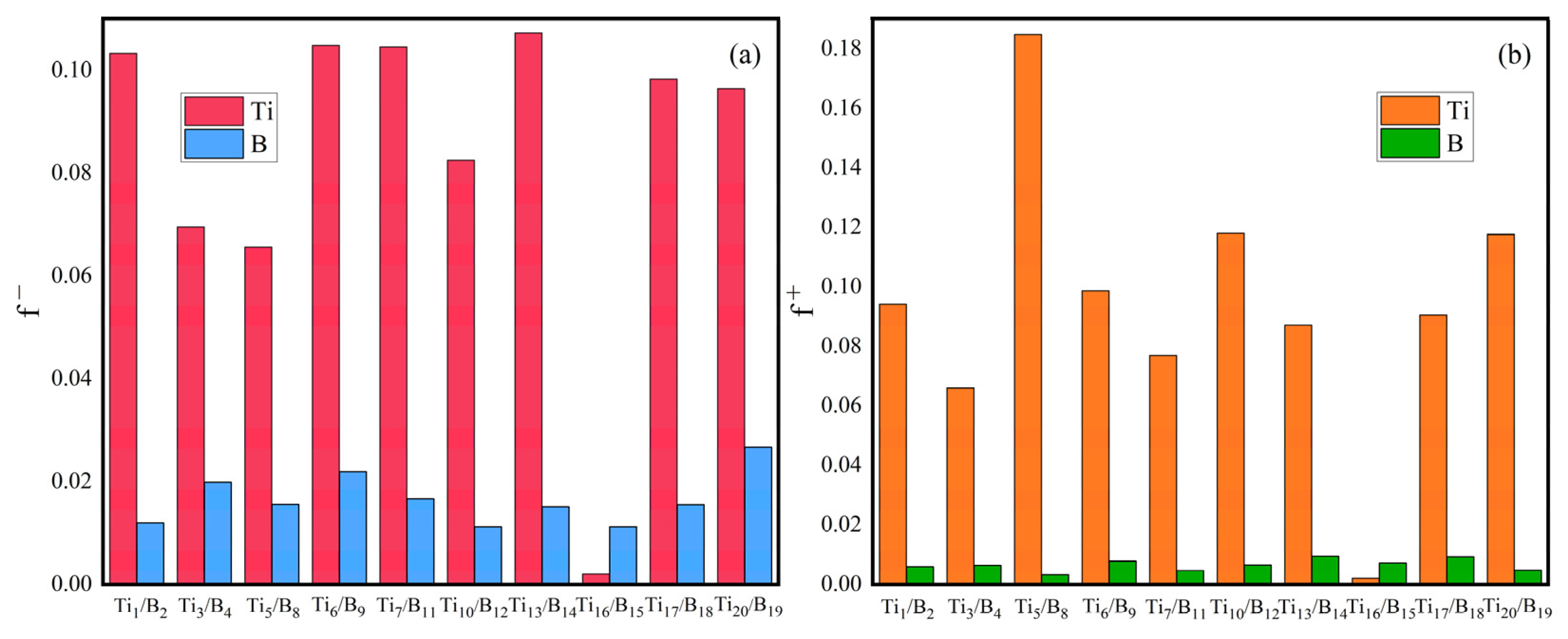
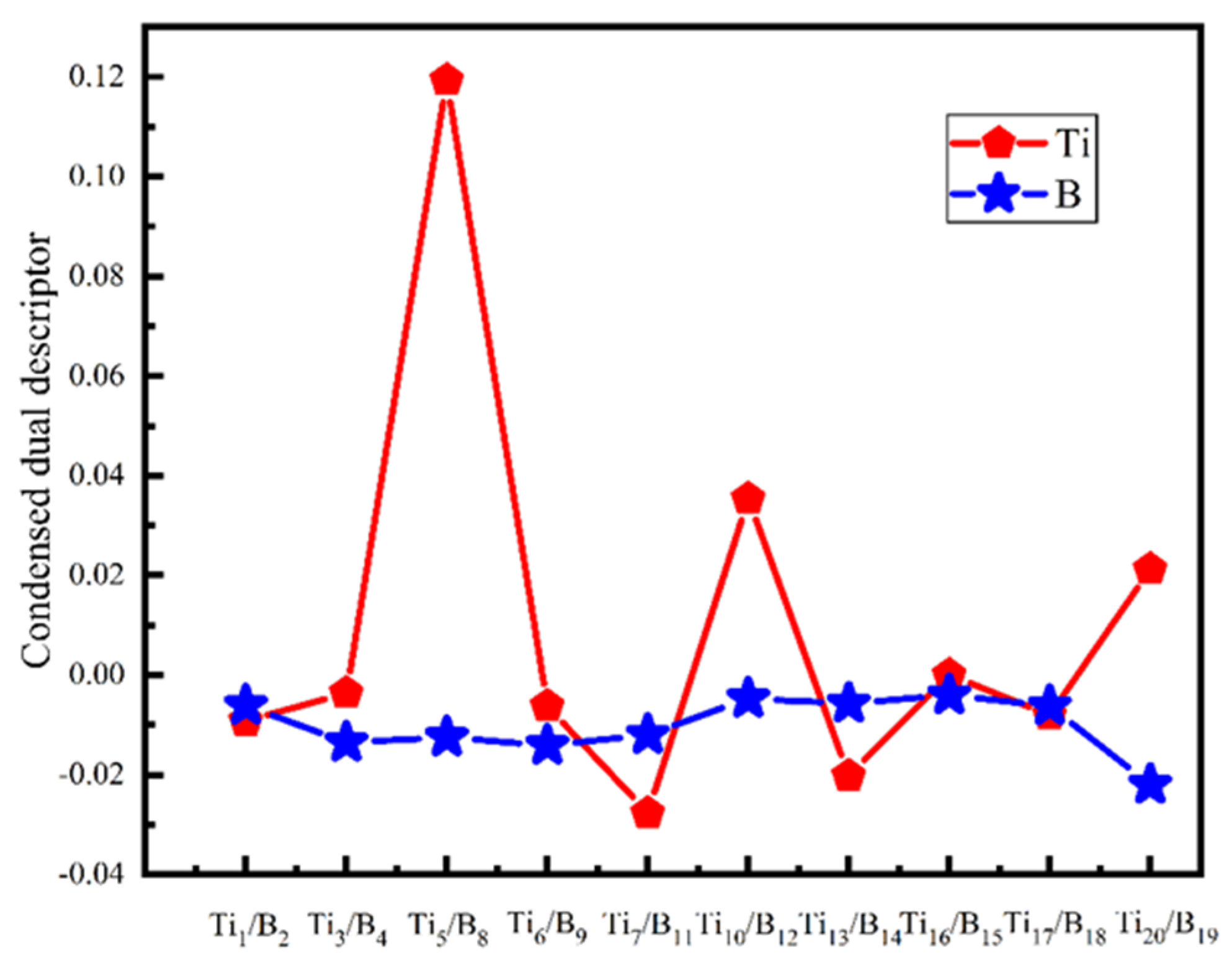
- It shows that HOMOs and LUMOs are concentrated on Ti atoms within the TiB2 structure, indicating that the TiB2 structure is the more active part of the clusters, and the activity of Ti atoms is higher than that of B atoms. The map of Fukui function shows that the Ti atoms are more reactive than B atoms. The value of the condensed Fukui function and condensed dual descriptor suggests that the value of most Ti atoms is much larger than that of B atoms. Therefore, Ti atoms have higher reactivity.
- (3)
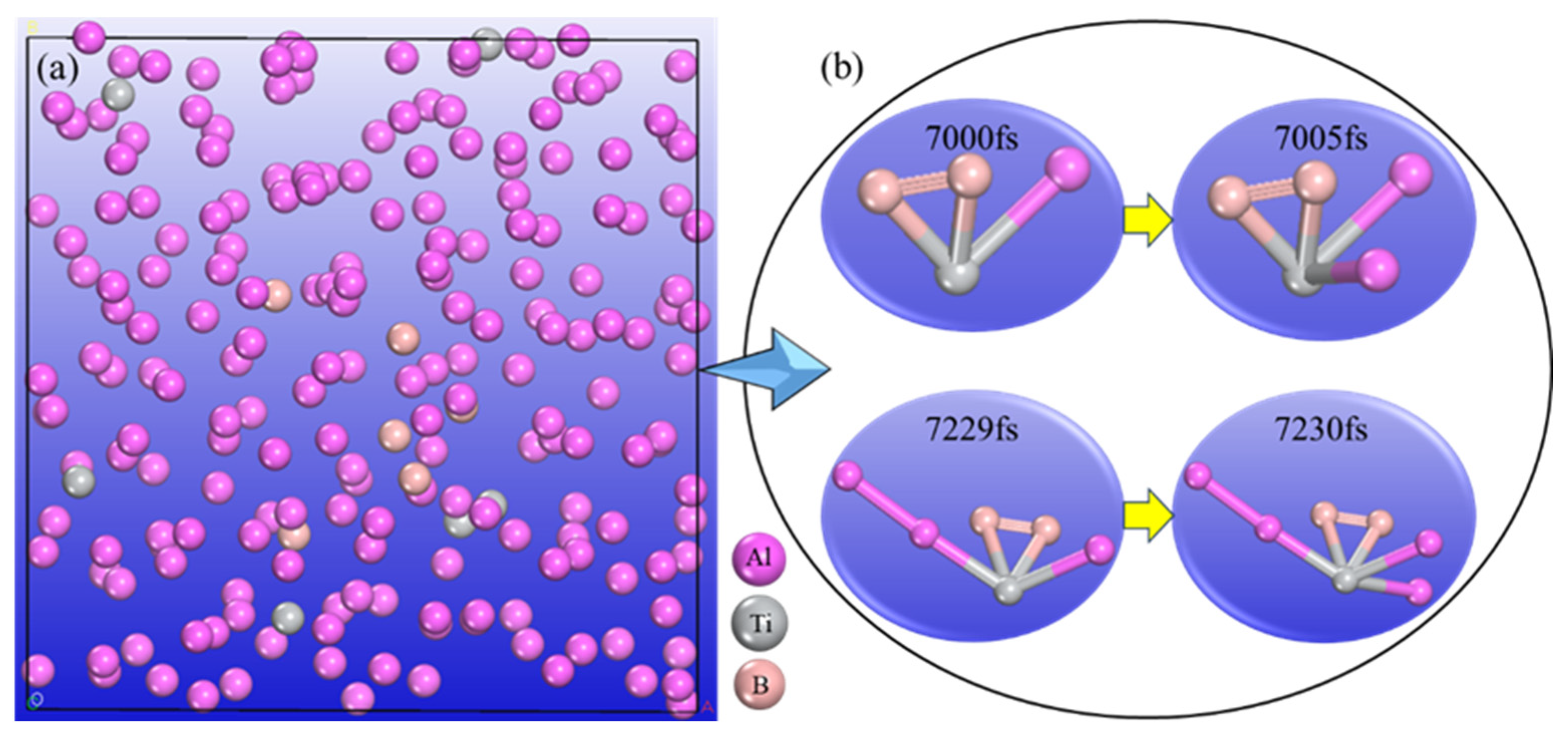
- The AIMD simulation results of the Al-Ti-B system show that Ti and B atoms can form TiB2 structure in situ, and there are Al atoms adsorbing and growing on the Ti surface in the TiB2 structure. The electronic structure of TiB2 was analysed by charge density difference. The results show that B atoms are connected by strong covalent bonds, and Ti and B atoms form ionic covalent bonds.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kori, S.; Murty, B.; Chakraborty, M. Development of an efficient grain refiner for Al–7Si alloy and its modification with strontium. Mater. Sci. Eng. A 2000, 283, 94–104. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Z.; Qiu, D.; Taylor, J.A.; Easton, M.A.; Zhang, M.-X. Revisiting the role of peritectics in grain refinement of Al alloys. Acta Mater. 2013, 61, 360–370. [Google Scholar] [CrossRef]
- Quested, T. Understanding mechanisms of grain refinement of aluminium alloys by inoculation. Mater. Sci. Technol. 2004, 20, 1357–1369. [Google Scholar] [CrossRef]
- Fan, Z.; Gao, F.; Wang, Y.; Men, H.; Zhou, L. Effect of solutes on grain refinement. Prog. Mater. Sci. 2022, 123, 100809. [Google Scholar] [CrossRef]
- Ding, W.; Zhao, X.; Chen, T.; Zhang, H.; Liu, X.; Cheng, Y.; Lei, D. Effect of rare earth Y and Al–Ti–B master alloy on the microstructure and mechanical properties of 6063 aluminum alloy. J. Alloys Compd. 2020, 830, 154685. [Google Scholar] [CrossRef]
- Murty, B.; Kori, S.; Chakraborty, M. Grain refinement of aluminium and its alloys by heterogeneous nucleation and alloying. Int. Mater. Rev. 2002, 47, 3–29. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Zhang, Y.; Qin, T.; Zhou, X.; Thompson, G.; Pennycook, T.; Hashimoto, T. Grain refining mechanism in the Al/Al–Ti–B system. Acta Mater. 2015, 84, 292–304. [Google Scholar] [CrossRef]
- Huang, B.; Liu, Y.; Zhou, Z.; Cheng, W.; Liu, X. Selective laser melting of 7075 aluminum alloy inoculated by Al–Ti–B: Grain refinement and superior mechanical properties. Vacuum 2022, 200, 111030. [Google Scholar] [CrossRef]
- Greer, A.L. Overview: Application of heterogeneous nucleation in grain-refining of metals. J. Chem. Phys. 2016, 145, 211704. [Google Scholar] [CrossRef]
- Mohanty, P.; Gruzleski, J. Grain refinement mechanisms of hypoeutectic Al Si alloys. Acta Mater. 1996, 44, 3749–3760. [Google Scholar] [CrossRef]
- Wang, J.; Horsfield, A.; Schwingenschlögl, U.; Lee, P.D. Heterogeneous nucleation of solid Al from the melt by TiB 2 and Al 3 Ti: An ab initio molecular dynamics study. Phys. Rev. B 2010, 82, 184203. [Google Scholar] [CrossRef]
- Yu, F.; Liu, Z.; Zhao, R.; Yang, J.; Qiao, J.; Hu, W. Effect of Al–Ti–B master alloy on microstructure and properties of aluminum-air battery anode materials. J. Mater. Res. Technol. 2023, 27, 4908–4919. [Google Scholar] [CrossRef]
- Jones, G.P.; Pearson, J. Factors affecting the grain-refinement of aluminum using titanium and boron additives. Metall. Trans. B 1976, 7, 223–234. [Google Scholar] [CrossRef]
- Sigworth, G.K.; Kuhn, T.A. Grain refinement of aluminum casting alloys. Int. J. Met. 2007, 1, 31–40. [Google Scholar] [CrossRef]
- Han, Y.; Liu, X.; Bian, X. In situ TiB2 particulate reinforced near eutectic Al–Si alloy composites. Compos. Part A Appl. Sci. Manuf. 2002, 33, 439–444. [Google Scholar] [CrossRef]
- Kumar, S.; Chakraborty, M.; Sarma, V.S.; Murty, B.S. Tensile and wear behaviour of in situ Al–7Si/TiB2 particulate composites. Wear 2008, 265, 134–142. [Google Scholar] [CrossRef]
- MAkbari, K.; Baharvandi, H.; Shirvanimoghaddam, K. Tensile and fracture behavior of nano/micro TiB2 particle reinforced casting A356 aluminum alloy composites. Mater. Des. 2015, 66, 150–161. [Google Scholar]
- Dong, B.-X.; Li, Q.; Wang, Z.-F.; Liu, T.-S.; Yang, H.-Y.; Shu, S.-L.; Chen, L.-Y.; Qiu, F.; Jiang, Q.-C.; Zhang, L.-C. Enhancing strength-ductility synergy and mechanisms of Al-based composites by size-tunable in-situ TiB2 particles with specific spatial distribution. Compos. Part B Eng. 2021, 217, 108912. [Google Scholar] [CrossRef]
- Li, P.; Li, Y.; Wu, Y.; Ma, G.; Liu, X. Distribution of TiB2 particles and its effect on the mechanical properties of A390 alloy. Mater. Sci. Eng. A 2012, 546, 146–152. [Google Scholar] [CrossRef]
- Greer, A.; Bunn, A.; Tronche, A.; Evans, P.; Bristow, D. Modelling of inoculation of metallic melts: Application to grain refinement of aluminium by Al–Ti–B. Acta Mater. 2000, 48, 2823–2835. [Google Scholar] [CrossRef]
- Feng, J.; Han, Y.; Han, X.; Wang, X.; Song, S.; Sun, B.; Chen, M.; Liu, P. Atomic insights into heterogeneous nucleation and growth kinetics of Al on TiB2 particles in undercooled Al-5Ti-1B melt. J. Mater. Sci. Technol. 2023, 156, 72–82. [Google Scholar] [CrossRef]
- Wearing, D.; Horsfield, A.P.; Xu, W.; Lee, P.D. Which wets TiB2 inoculant particles: Al or Al3Ti? J. Alloys Compd. 2016, 664, 460–468. [Google Scholar] [CrossRef]
- Liu, X.; Wang, B.; Li, Q.; Wang, J.; Zhang, C.; Xue, C.; Yang, X.; Tian, G.; Liu, X.; Tang, H. Quantifying the effects of grain refiners Al-Ti-B and La on the microstructure and mechanical properties of W319 alloy. Metals 2022, 12, 627. [Google Scholar] [CrossRef]
- Knaislová, A.; Michna, Š.; Hren, I.; Vlach, T.; Michalcová, A.; Novák, P.; Stančeková, D. Microstructural characteristics of Al-Ti-B inoculation wires and their addition to the AlSi7Mg0.3 alloy. Materials 2022, 15, 7626. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wang, H.; Gao, Z.; Wu, Y.; Wang, M.; Jin, X.; Leung, C.L.A.; Lee, P.; Fu, Y.; Wang, H. The role of in-situ nano-TiB2 particles in improving the printability of noncastable 2024Al alloy. Mater. Res. Lett. 2022, 10, 656–665. [Google Scholar] [CrossRef]
- Youssef, Y.; Dashwood, R.; Lee, P. Effect of clustering on particle pushing and solidification behaviour in TiB2 reinforced aluminium PMMCs. Compos. Part A Appl. Sci. Manuf. 2005, 36, 747–763. [Google Scholar] [CrossRef]
- Wang, M.; Chen, D.; Chen, Z.; Wu, Y.; Wang, F.; Ma, N.; Wang, H. Mechanical properties of in-situ TiB2/A356 composites. Mater. Sci. Eng. A 2014, 590, 246–254. [Google Scholar] [CrossRef]
- Xi, L.; Gu, D.; Guo, S.; Wang, R.; Ding, K.; Prashanth, K.G. Grain refinement in laser manufactured Al-based composites with TiB2 ceramic. J. Mater. Res. Technol. 2020, 9, 2611–2622. [Google Scholar] [CrossRef]
- Xiao, Y.; Bian, Z.; Wu, Y.; Ji, G.; Li, Y.; Li, M.; Lian, Q.; Chen, Z.; Addad, A.; Wang, H. Effect of nano-TiB2 particles on the anisotropy in an AlSi10Mg alloy processed by selective laser melting. J. Alloys Compd. 2019, 798, 644–655. [Google Scholar] [CrossRef]
- Guo, X.; Li, X.; Li, Y.; Yang, J.; Wan, X.; Chen, L.; Liu, J.; Liu, X.; Yu, R.; Zheng, L. Molecule template method for precise synthesis of Mo-based alloy clusters and electrocatalytic nitrogen reduction on partially reduced PtMo alloy oxide cluster. Nano Energy 2020, 78, 105211. [Google Scholar] [CrossRef]
- Kurashige, W.; Hayashi, R.; Wakamatsu, K.; Kataoka, Y.; Hossain, S.; Iwase, A.; Kudo, A.; Yamazoe, S.; Negishi, Y. Atomic-level understanding of the effect of heteroatom doping of the cocatalyst on water-splitting activity in AuPd or AuPt alloy cluster-loaded BaLa4Ti4O15. ACS Appl. Energy Mater. 2019, 2, 4175–4187. [Google Scholar] [CrossRef]
- Rodríguez-Kessler, P.; Muñoz-Castro, A.; Alonso-Dávila, P.; Aguilera-Granja, F.; Rodríguez-Domínguez, A. Structural, electronic and catalytic properties of bimetallic PtnAgn (n = 1–7) clusters. J. Alloys Compd. 2020, 845, 155897. [Google Scholar] [CrossRef]
- Huang, H.; Wu, B.; Gao, Q.; Li, P.; Yang, X. Structural, electronic and spectral properties referring to hydrogen storage capacity in binary alloy ScBn (n= 1–12) clusters. Int. J. Hydrogen Energy 2017, 42, 21086–21095. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, L. Structural evolution and electronic properties of Cu-Zn alloy clusters. J. Alloys Compd. 2019, 771, 762–768. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16, Revision A. 03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. Realization of Conceptual Density Functional Theory and Information-Theoretic Approach in Multiwfn Program. In Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory; Wiley: Hoboken, NJ, USA, 2022; Volume 2, pp. 631–647. [Google Scholar]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0. 16; Semichem Inc.: Shawnee Mission, KS, USA, 2016; pp. 143–150. [Google Scholar]
- Hafner, J.; Kresse, G. The vienna ab-initio simulation program VASP: An efficient and versatile tool for studying the structural, dynamic, and electronic properties of materials. In Properties of Complex Inorganic Solids; Springer: Berlin/Heidelberg, Germany, 1997; pp. 69–82. [Google Scholar]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef]
- Fujinaga, T.; Watanabe, Y.; Shibuta, Y. Nucleation dynamics in Al solidification with Al-Ti refiners by molecular dynamics simulation. Comput. Mater. Sci. 2020, 182, 109763. [Google Scholar] [CrossRef]
- Zhang, H.; Han, Y.; Dai, Y.; Lu, S.; Wang, J.; Zhang, J.; Shu, D.; Sun, B. An ab initio study on the electronic structures of the solid/liquid interface between TiB2 (0 0 0 1) surface and Al melts. J. Alloys Compd. 2014, 615, 863–867. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, F.; van Speybroeck, V.; Waroquier, M.; Geerlings, P.; de Proft, F. Electrophilicity and nucleophilicity index for radicals. Org. Lett. 2007, 9, 2721–2724. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Maiti, B.; Sarkar, U. Philicity: A unified treatment of chemical reactivity and selectivity. J. Phys. Chem. A 2003, 107, 4973–4975. [Google Scholar] [CrossRef]
- Oláh, J.; van Alsenoy, C.; Sannigrahi, A. Condensed Fukui functions derived from stockholder charges: Assessment of their performance as local reactivity descriptors. J. Phys. Chem. A 2002, 106, 3885–3890. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why is the dual descriptor a more accurate local reactivity descriptor than Fukui functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Beck, M.E. Do Fukui function maxima relate to sites of metabolism? A critical case study. J. Chem. Inf. Model. 2005, 45, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Giri, S. Electrophilicity index within a conceptual DFT framework. Annu. Rep. Sect. C Phys. Chem. 2009, 105, 13–39. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Fuentealba, P.; Pérez, P. Contreras, On the condensed Fukui function. J. Chem. Phys. 2000, 133, 2544–2551. [Google Scholar] [CrossRef]
- Han, Y.; Dai, Y.; Shu, D.; Wang, J.; Sun, B. First-principles calculations on the stability of Al/Ti B 2 interface. Appl. Phys. Lett. 2006, 89, 144107. [Google Scholar] [CrossRef]
- Edim, M.M.; Enudi, O.C.; Asuquo, B.B.; Louis, H.; Bisong, E.A.; Agwupuye, J.A.; Chioma, A.G.; Odey, J.O.; Joseph, I.; Bassey, F.I. Aromaticity indices, electronic structural properties, and fuzzy atomic space investigations of naphthalene and its aza-derivatives. Heliyon 2021, 7, e06138. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Ren, Q.; Chen, F.; Lu, T. Comparative study on the methods for predicting the reactive site of nucleophilic reaction. Sci. China Chem. 2015, 58, 1845–1852. [Google Scholar] [CrossRef]
- Pino-Rios, R.; Inostroza, D.; Cárdenas-Jirón, G.; Tiznado, W. Orbital-weighted dual descriptor for the study of local reactivity of systems with (quasi-) degenerate states. J. Phys. Chem. A 2019, 123, 10556–10562. [Google Scholar] [CrossRef]
- Fievez, T.; Sablon, N.; de Proft, F.; Ayers, P.W.; Geerlings, P. Calculation of Fukui functions without differentiating to the number of electrons. 3. Local Fukui function and dual descriptor. J. Chem. Theory Comput. 2008, 4, 1065–1072. [Google Scholar] [CrossRef]
- Zhang, H.; Han, Y.; Dai, Y.; Wang, J.; Sun, B. An ab initio molecular dynamics study: Liquid-Al/solid-TiB2 interfacial structure during heterogeneous nucleation. J. Phys. D Appl. Phys. 2012, 45, 455307. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Ground State Structures of TinBn (n = 2–12) Clusters | ||
|---|---|---|---|
 |  |  |  |
| Ti2B2(C1) | Ti3B3(C1) | Ti4B4(C1) | Ti5B5(C1) |
 |  |  | 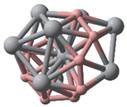 |
| Ti6B6(C1) | Ti7B7(C1) | Ti8B8(C1) | Ti9B9(C1) |
 |  |  |  |
| Ti10B10(C1) | Ti11B11(C1) | Ti12B12(C1) | |
| Categories | Ground State Structures of TinBn (n = 2–12) Clusters | |||
|---|---|---|---|---|
| HOMO | 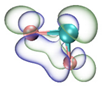 | 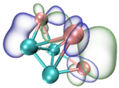 |  | 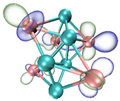 |
| LUMO |  | 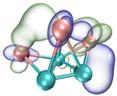 | 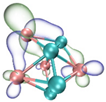 | 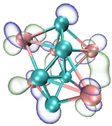 |
| n | 2 | 3 | 4 | 5 |
| HOMO | 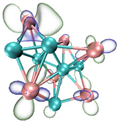 | 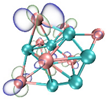 | 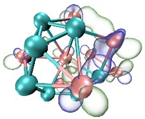 | 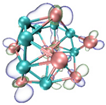 |
| LUMO | 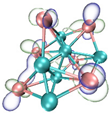 | 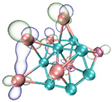 | 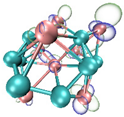 | 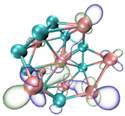 |
| n | 6 | 7 | 8 | 9 |
| HOMO | 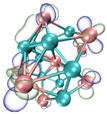 | 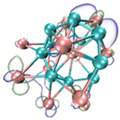 | 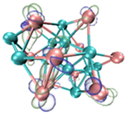 |  ---Ti ---Ti |
| LUMO | 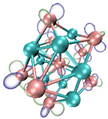 | 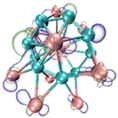 | 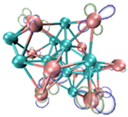 |  ---B ---B |
| n | 10 | 11 | 12 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Zhang, H.; Zhao, X.; Liu, B.; Chen, X.; Zhou, L. Study on the Microscopic Mechanism of the Grain Refinement of Al-Ti-B Master Alloy. Metals 2024, 14, 197. https://doi.org/10.3390/met14020197
Yang L, Zhang H, Zhao X, Liu B, Chen X, Zhou L. Study on the Microscopic Mechanism of the Grain Refinement of Al-Ti-B Master Alloy. Metals. 2024; 14(2):197. https://doi.org/10.3390/met14020197
Chicago/Turabian StyleYang, Lianfeng, Huan Zhang, Xiran Zhao, Bo Liu, Xiumin Chen, and Lei Zhou. 2024. "Study on the Microscopic Mechanism of the Grain Refinement of Al-Ti-B Master Alloy" Metals 14, no. 2: 197. https://doi.org/10.3390/met14020197
APA StyleYang, L., Zhang, H., Zhao, X., Liu, B., Chen, X., & Zhou, L. (2024). Study on the Microscopic Mechanism of the Grain Refinement of Al-Ti-B Master Alloy. Metals, 14(2), 197. https://doi.org/10.3390/met14020197