
Influence of Th, Zr, and Ti Dopants on Solution Property of Xe in Uranium Dioxide with Defects: A DFT + U Study

 ,
,
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Model Verification
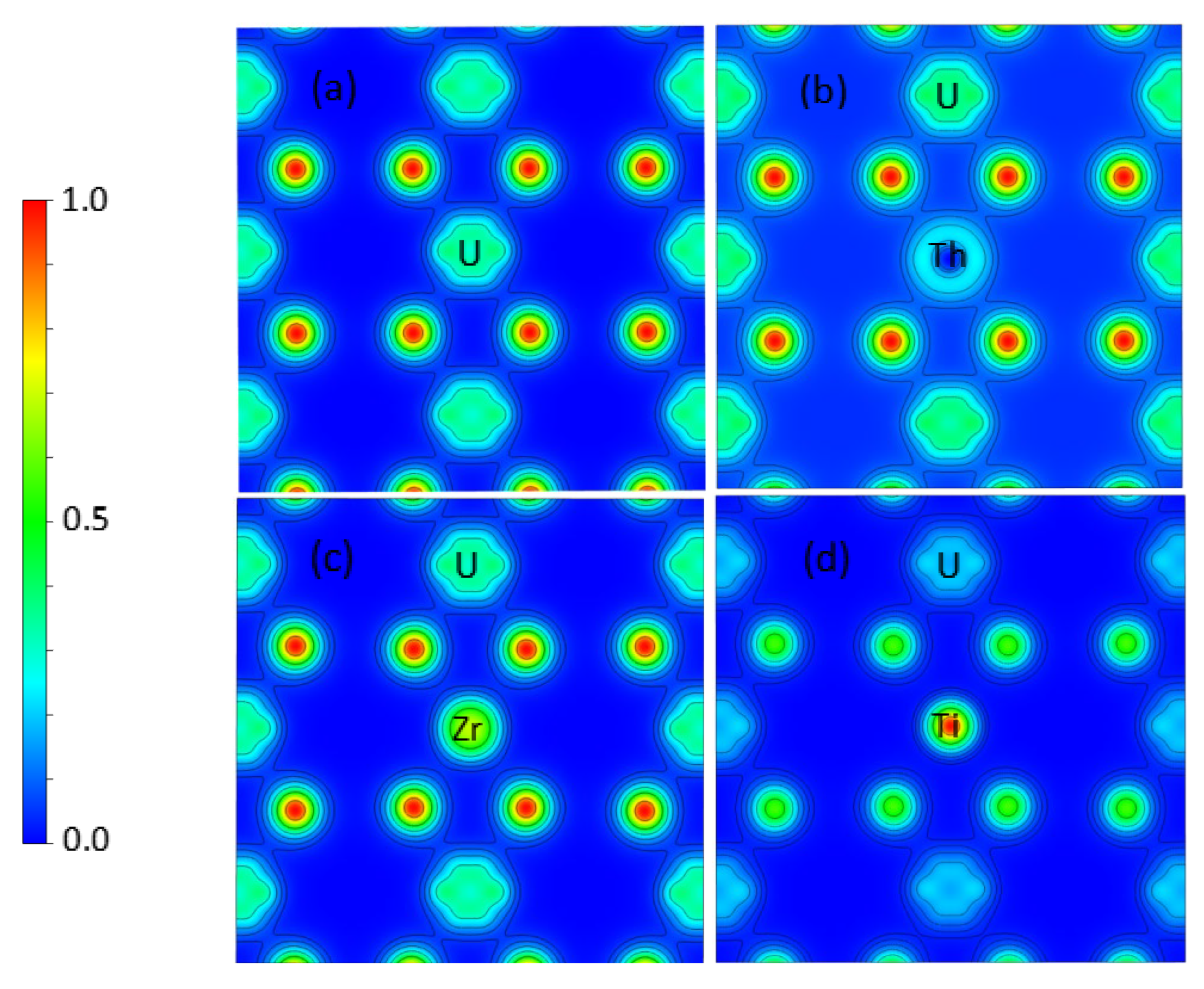
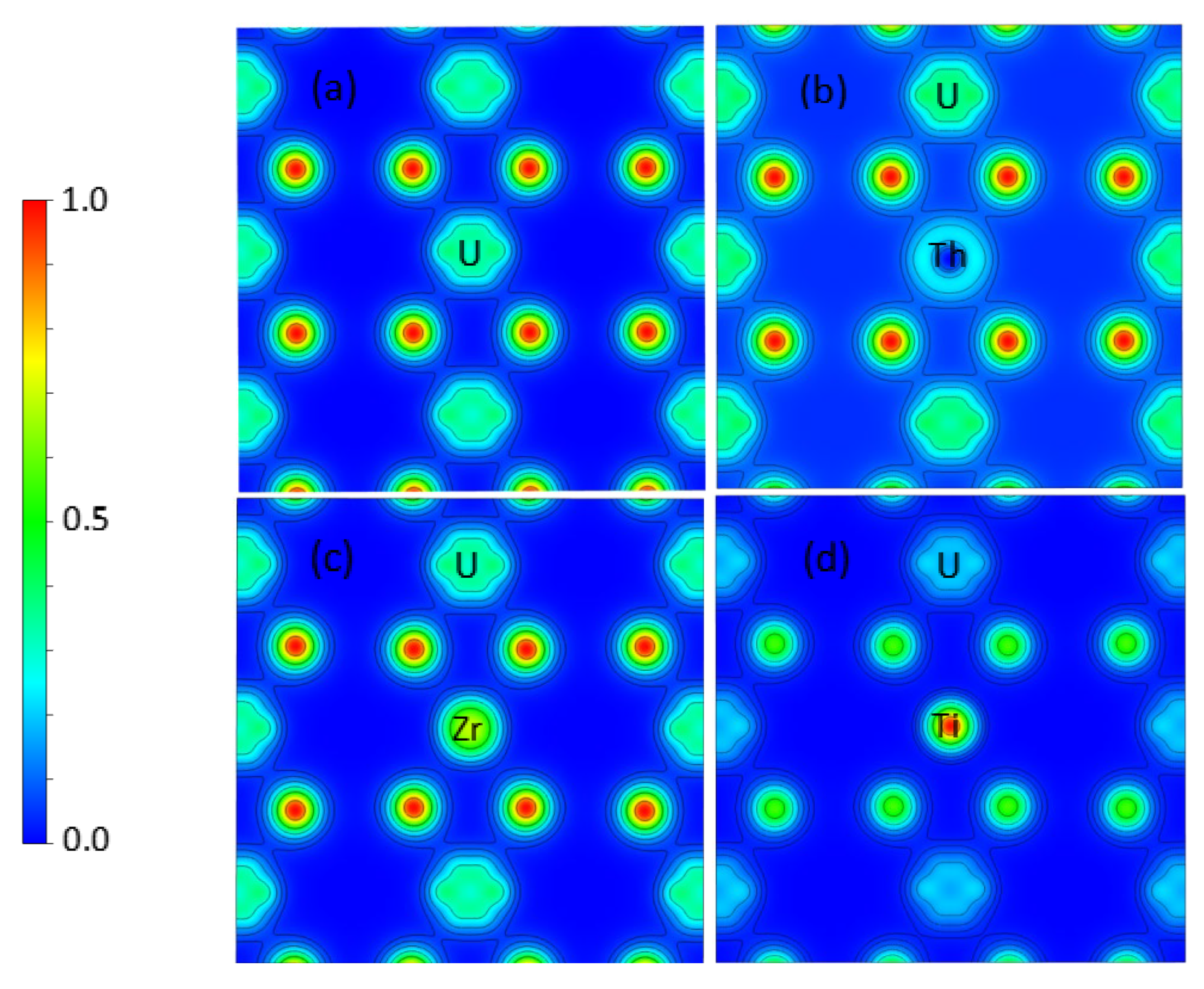
3.2. Th, Zr, and Ti Dopants in UO2
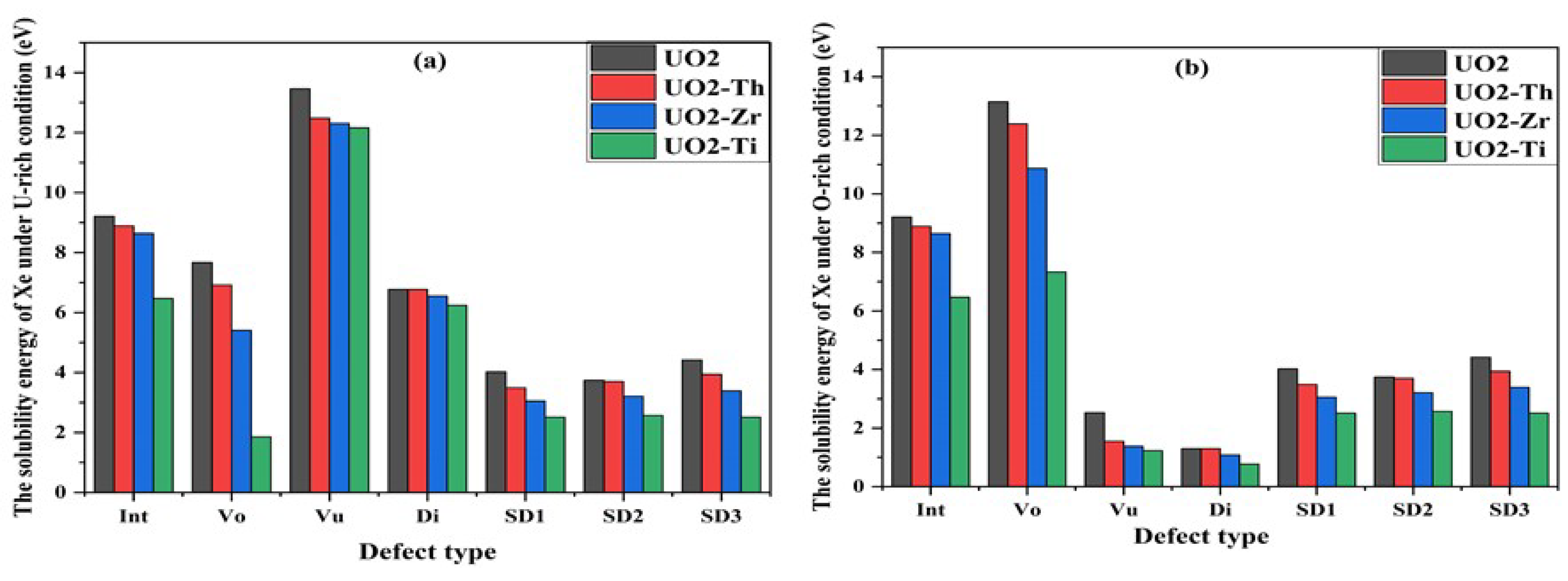
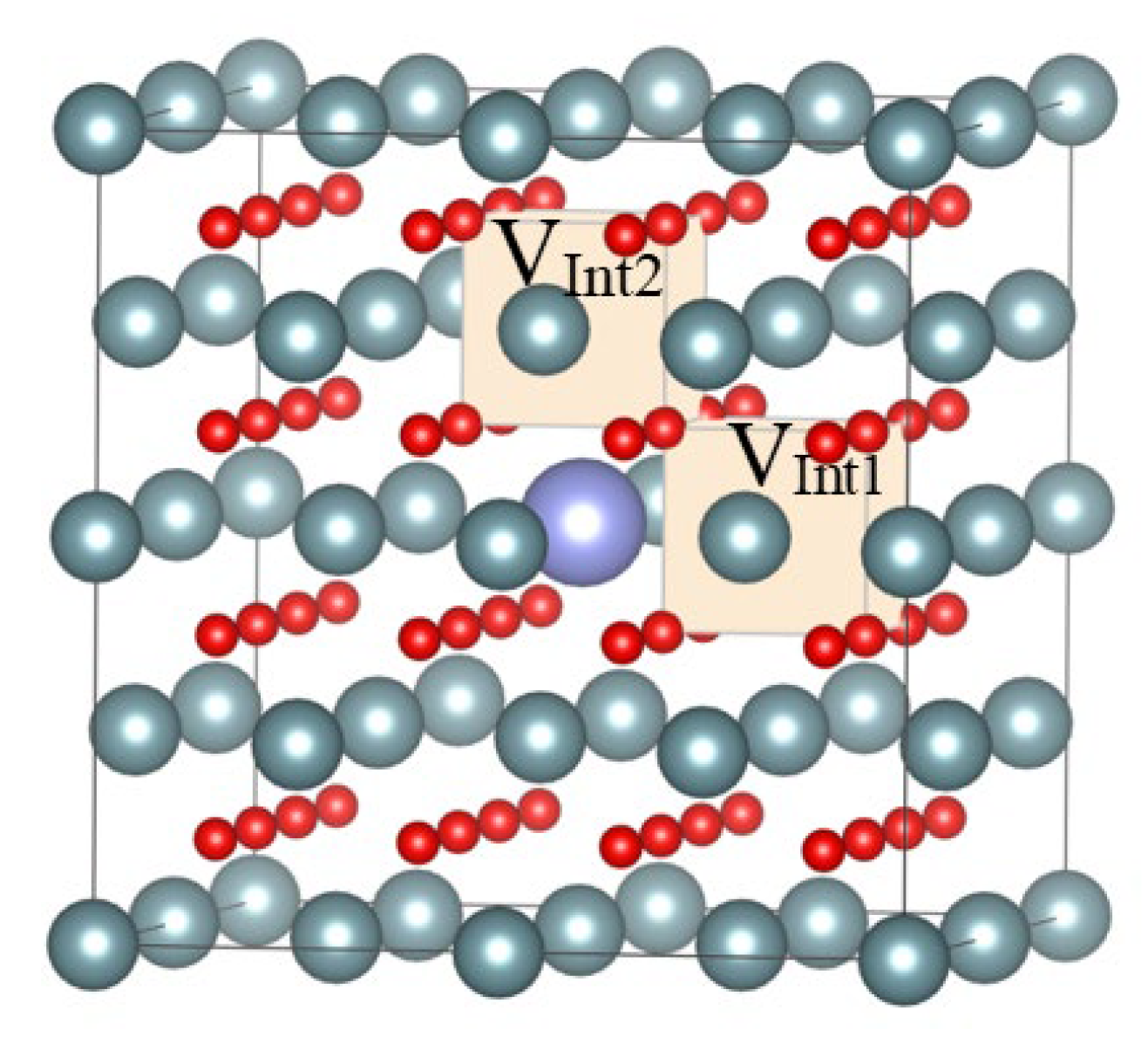
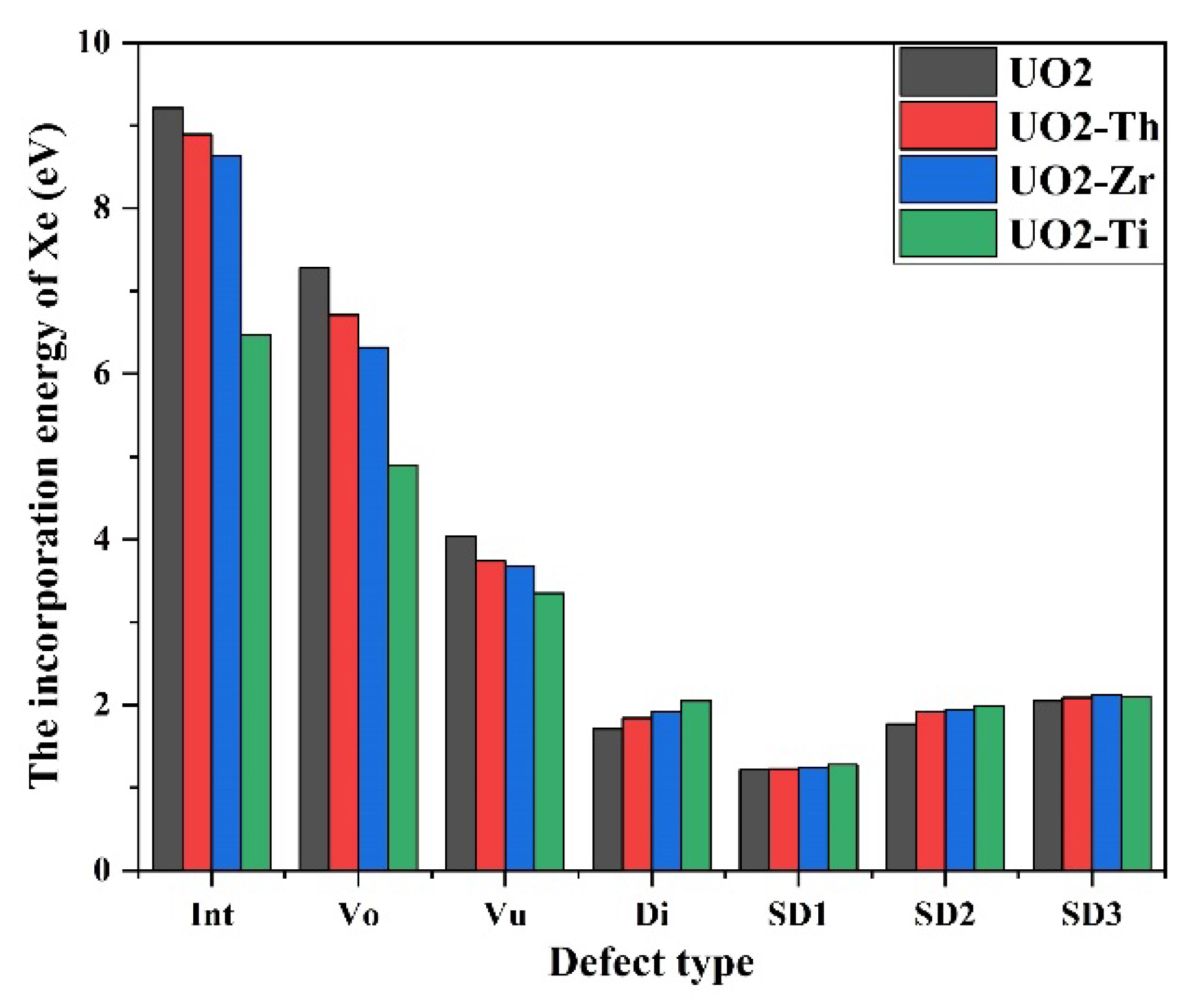
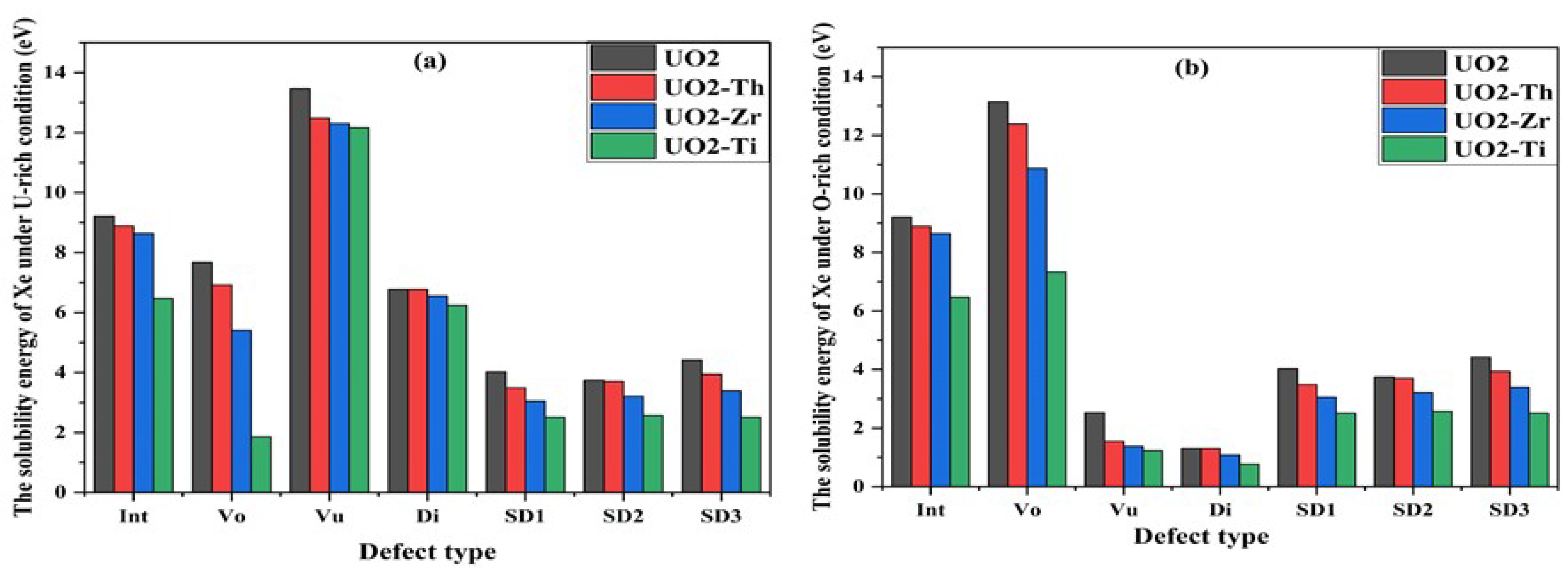
3.3. Effect of Dopant on Xe in UO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.-J.; Lan, J.-H.; Wang, C.-Z.; Wu, Q.-Y.; Bo, T.; Chai, Z.-F.; Shi, W.-Q. Theoretical Investigation on Incorporation and Diffusion Properties of Xe in Uranium Mononitride. J. Phys. Chem. C 2015, 119, 5783–5789. [Google Scholar] [CrossRef]
- Andersson, A.D.; Perriot, R.T.; Pastore, G.; Tonks, M.R.; Cooper, M.W.; Liu, X.-Y.; Goyal, A.; Uberuaga, B.P.; Stanek, C.R. Report on Simulation of Fission Gas and Fission Product Diffusion in Uo2; Los Alamos National Lab.: Los Alamos, NM, USA, 2016. [Google Scholar] [CrossRef]
- White, R.J.; Tucker, M.O. A new fission-gas release model. J. Nucl. Mater. 1983, 118, 1–38. [Google Scholar] [CrossRef]
- White, R.J. The development of grain-face porosity in irradiated oxide fuel. J. Nucl. Mater. 2004, 325, 61–77. [Google Scholar] [CrossRef]
- B.RT.Frost. Volumes 10A and 10B of Materials science and technology–A Comprehensive treatment. Adv. Mater. 1995, 7, 1047. [Google Scholar] [CrossRef]
- Zinkle, S.J.; Was, G.S. Materials challenges in nuclear energy. Acta Mater. 2013, 61, 735–758. [Google Scholar] [CrossRef]
- Pang, J.W.L.; Chernatynskiy, A.; Larson, B.C.; Buyers, W.J.L.; Abernathy, D.L.; McClellan, K.J.; Phillpot, S.R. Phonon density of states and anharmonicity of UO2. Phys. Rev. B 2014, 89, 115132. [Google Scholar] [CrossRef] [Green Version]
- Ao, B.; Lu, H. First-principles energetics of rare gases incorporation into uranium dioxide. Phys. Rev. B 2016, 373, 102–109. [Google Scholar] [CrossRef]
- Une, K.; Tanabe, I.; Oguma, M. Effects of additives and the oxygen potential on the fission gas diffusion in UO2 fuel. J. Nucl. Mater. 1987, 150, 93–99. [Google Scholar] [CrossRef]
- Fujino, T.; Shiratori, T.; Sato, N.; Fukuda, K.; Yamada, K.; Serizawa, H. Post-irradiation examination of high burnup Mg doped UO2 in comparison with undoped UO2, Mg–Nb doped UO2 and Ti doped UO2. J. Nucl. Mater. 2001, 297, 176–205. [Google Scholar] [CrossRef]
- Fujino, T.; Nakama, S.; Sato, N.; Yamada, K.; Fukuda, K.; Serizawa, H.; Shiratori, T. Solubility of magnesium in uranium dioxide. J. Nucl. Mater. 1997, 246, 150–157. [Google Scholar] [CrossRef]
- Kashibe, S.; Une, K. Effect of additives (Cr2O3, Al2O3, SiO2, MgO) on diffusional release of 133Xe from UO2 fuels. J. Nucl. Mater. 1998, 254, 234–242. [Google Scholar] [CrossRef]
- Matsui, T.; Arita, Y.; Naito, K. High temperature heat capacities and electrical conductivities of UO2 doped with Ti, Nb and Sc. Solid State Ion. 1991, 49, 195–200. [Google Scholar] [CrossRef]
- Aybers, M.T.; Artir, R.; Akşit, A.A.; Akbal, S. Investigation of some mechanical properties of Ti2O3-doped UO2 fuel pellets. Mater. Charact. 2006, 57, 182–186. [Google Scholar] [CrossRef]
- Peres, V.; Favergeon, L.; Andrieu, M.; Palussière, J.C.; Balland, J.; Delafoy, C.; Pijolat, M. High temperature chromium volatilization from Cr2O3 powder and Cr2O3-doped UO2 pellets in reducing atmospheres. J. Nucl. Mater. 2012, 423, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Shen, L.; Yang, G.-W.; Li, Q.-Y.; Zhu, L.-L.; Shen, W.; Ji, C.; Shen, X.-F. Four novel coordination compounds containing Hpypza ligand [Hpypza=[3-(2-pyridyl)-1-pyrazolyl] acetic acid]. Inorg. Chim. Acta 2012, 392, 25–29. [Google Scholar] [CrossRef]
- Cohen, I.; Schaner, B.E. A metallographic and X-ray study of the UO2-ZrO2 system. J. Nucl. Mater. 1963, 9, 18–52. [Google Scholar] [CrossRef]
- Kulkarni, N.K.; Krishnan, K.; Kasar, U.M.; Rakshit, S.K.; Sali, S.K.; Aggarwal, S.K. Thermal studies on fluorite type ZryU1−yO2 solid solutions. J. Nucl. Mater. 2009, 384, 81–86. [Google Scholar] [CrossRef]
- Xiao, H.; Long, C.; Chen, H.; Tian, X.; Wei, T.; Zhao, Y.; Gao, W. Effects of Zr doping on the surface energy and surface structure of UO2: Atomistic simulations. Appl. Surf. Sci. 2015, 351, 517–523. [Google Scholar] [CrossRef]
- Ang, C.Y.; Burkhammer, K.W. Sintering of high density uranium dioxide bodies. J. Nucl. Mater. 1960, 2, 176–180. [Google Scholar] [CrossRef]
- Rickert, K.; Prusnick, T.A.; Hunt, E.; Kimani, M.M.; Chastang, S.; Brooks, D.L.; Moore, E.A.; Petrosky, J.C.; Mann, J.M. Inhibiting laser oxidation of UO2 via Th substitution. J. Nucl. Mater. 2019, 517, 254–262. [Google Scholar] [CrossRef]
- Sholl, D.; Steckel, J.A. Density Functional Theory: A Practical Introduction; John Wiley & Sons: New York, NY, USA, 2011. [Google Scholar]
- Hohenberg, P.; Kohn, W. Density functional theory (DFT). Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Ao, B.; Qiu, R.; Zhang, G.; Pu, Z.; Wang, X.; Shi, P. Light impurity atoms as the probes for the electronic structures of actinide dioxides. Comput. Mater. Sci. 2018, 142, 25–31. [Google Scholar] [CrossRef]
- Van Brutzel, L.; Rarivomanantsoa, M. Molecular dynamics simulation study of primary damage in UO2 produced by cascade overlaps. J. Nucl. Mater. 2006, 358, 209–216. [Google Scholar] [CrossRef]
- Martin, G.; Garcia, P.; Van Brutzel, L.; Dorado, B.; Maillard, S. Effect of the cascade energy on defect production in uranium dioxide. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2011, 269, 1727–1730. [Google Scholar] [CrossRef]
- Yu, J.; Devanathan, R.; Weber, W.J. First-principles study of defects and phase transition in UO2. J. Phys. Condens. Matter 2009, 21, 435401. [Google Scholar] [CrossRef]
- Nerikar, P.; Watanabe, T.; Tulenko, J.S.; Phillpot, S.R.; Sinnott, S.B. Energetics of intrinsic point defects in uranium dioxide from electronic-structure calculations. J. Nucl. Mater. 2009, 384, 61–69. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Dorado, B.; Freyss, M.; Amadon, B.; Bertolus, M.; Jomard, G.; Garcia, P. Advances in first-principles modelling of point defects in UO2: f electron correlations and the issue of local energy minima. J. Phys. Condens. Matter 2013, 25, 333201. [Google Scholar] [CrossRef] [PubMed]
- Jollet, F.; Petit, T.; Gota, S.; Thromat, N.; Gautier-Soyer, M.; Pasturel, A. The electronic structure of uranium dioxide: An oxygen K-edge X-ray absorption study. J. Phys. Condens. Matter 1997, 9, 9393–9401. [Google Scholar] [CrossRef]
- Crocombette, J.P.; Jollet, F.; Nga, L.T.; Petit, T. Plane-wave pseudopotential study of point defects in uranium dioxide. Phys. Rev. B 2001, 64, 104107. [Google Scholar] [CrossRef] [Green Version]
- Freyss, M.; Petit, T.; Crocombette, J.-P. Point defects in uranium dioxide: Ab initio pseudopotential approach in the generalized gradient approximation. J. Nucl. Mater. 2005, 347, 44–51. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Castell, M.R.; Botton, G.A.; Savrasov, S.Y.; Muggelberg, C.; Briggs, G.A.D.; Sutton, A.P.; Goddard, D.T. Understanding STM images and EELS spectra of oxides with strongly correlated electrons: A comparison of nickel and uranium oxides. Micron 2000, 31, 363–372. [Google Scholar] [CrossRef]
- Dorado, B.; Amadon, B.; Freyss, M.; Bertolus, M. DFT+U calculations of the ground state and metastable states of uranium dioxide. Phys. Rev. B 2009, 79, 235125. [Google Scholar] [CrossRef]
- Solomon, J.M.; Alexandrov, V.; Sadigh, B.; Navrotsky, A.; Asta, M. Computational study of the energetics and defect clustering tendencies for Y- and La-doped UO2. Acta Mater. 2014, 78, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Dorado, B.; Jomard, G.; Freyss, M.; Bertolus, M. Stability of oxygen point defects in UO2 by first-principles DFT+U calculations: Occupation matrix control and Jahn-Teller distortion. Phys. Rev. B 2010, 82, 035114. [Google Scholar] [CrossRef]
- Grimes, R.W.; Catlow, C.R.A. The stability of fission products in uranium dioxide. Philos. Trans. R. Soc. London. Ser. A Phys. Eng. Sci. 1991, 335, 609–634. [Google Scholar] [CrossRef]
- Idiri, M.; Le Bihan, T.; Heathman, S.; Rebizant, J. Behavior of actinide dioxides under pressure: UO2 and ThO2. Phys. Rev. B 2004, 70. [Google Scholar] [CrossRef]
- Torres, E.; Kaloni, T.P. Thermal conductivity and diffusion mechanisms of noble gases in uranium dioxide: A DFT+U study. J. Nucl. Mater. 2019, 521, 137–145. [Google Scholar] [CrossRef]
- Pegg, J.T.; Aparicio-Anglès, X.; Storr, M.; de Leeuw, N.H. DFT+U study of the structures and properties of the actinide dioxides. J. Nucl. Mater. 2017, 492, 269–278. [Google Scholar] [CrossRef]
- Gupta, F.; Brillant, G.; Pasturel, A. Correlation effects and energetics of point defects in uranium dioxide: A first principle investigation. Philos. Mag. 2007, 87, 2561–2569. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-W.; Tobin, J.G.; Crowhurst, J.C.; Sharma, S.; Dewhurst, J.K.; Velasco, P.O.; Yang, W.L.; Siekhaus, W.J. f-f origin of the insulating state in uranium dioxide: X-ray absorption experiments and first-principles calculations. Phys. Rev. B 2011, 83, 165102. [Google Scholar] [CrossRef]
- Faber, J.; Lander, G.H. Neutron diffraction study of UO2: Antiferromagnetic state. Phys. Rev. B 1976, 14, 1151–1164. [Google Scholar] [CrossRef]
- Fritz, I.J. Elastic properties of UO2 at high pressure. J. Appl. Phys. 1976, 47, 4353–4358. [Google Scholar] [CrossRef]
- Brooks, M.S.S.; Kelly, P.J. On the cohesive energy and charge density of uranium dioxide. Solid State Commun. 1983, 45, 689–692. [Google Scholar] [CrossRef]
- Lan, J.-H.; Wang, L.; Li, S.; Yuan, L.-Y.; Feng, Y.-X.; Sun, W.; Zhao, Y.-L.; Chai, Z.-F.; Shi, W.-Q. First principles modeling of zirconium solution in bulk UO2. J. Appl. Phys. 2013, 113, 183514. [Google Scholar] [CrossRef]
- Matzke, H.J. Gas release mechanisms in UO2—A critical review. Radiat. Eff. 1980, 53, 219–242. [Google Scholar] [CrossRef]
- Bès, R.; Martin, P.; Vathonne, E.; Delorme, R.; Sabathier, C.; Freyss, M.; Bertolus, M.; Glatzel, P. Experimental evidence of Xe incorporation in Schottky defects in UO2. Appl. Phys. Lett. 2015, 106, 114102. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, T.R.; Dixon, J. Electron irradiation of single crystal thorium dioxide and electron transfer reactions. Inorg. Chim. Acta 2000, 300–302, 305–313. [Google Scholar] [CrossRef]
- Shields, A.; Santos-Carballal, D.; de Leeuw, N.H. A density functional theory study of uranium-doped thoria and uranium adatoms on the major surfaces of thorium dioxide. J. Nucl. Mater. 2016, 473, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Delarmelina, M.; Quesne, M.G.; Catlow, C.R.A. Modelling the bulk properties of ambient pressure polymorphs of zirconia. Phys. Chem. Chem. Phys. 2020, 22, 6660–6676. [Google Scholar] [CrossRef] [PubMed]
- Stausholm-Møller, J.; Kristoffersen, H.H.; Hinnemann, B.; Madsen, G.K.; Hammer, B. DFT+ U study of defects in bulk rutile TiO2. J. Chem. Phys. 2010, 133, 144708. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.J.; Watson, G.W. Intrinsic n-type Defect Formation in TiO2: A Comparison of Rutile and Anatase from GGA+U Calculations. J. Phys. Chem. C 2010, 114, 2321–2328. [Google Scholar] [CrossRef]
- Morgan, B.; Watson, G. A DFT+U description of oxygen vacancies at the TiO2 rutile (110) surface. Surf. Sci. 2007, 601, 5034–5041. [Google Scholar] [CrossRef]
- Prada, S.; Rosa, M.; Giordano, L.; Di Valentin, C.; Pacchioni, G. Density functional theory study of TiO2/Ag interfaces and their role in memristor devices. Phys. Rev. B 2011, 83. [Google Scholar] [CrossRef]
- Morgan, B.J.; Madden, P.A. Lithium intercalation into TiO2(B): A comparison of LDA, GGA, and GGA+Udensity functional calculations. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef]
- Olsen, J.S.; Gerward, L.; Kanchana, V.; Vaitheeswaran, G. The bulk modulus of ThO2—an experimental and theoretical study. J. Alloy. Compd. 2004, 381, 37–40. [Google Scholar] [CrossRef]
- Idiri, M.; Le Bihan, T.; Heathman, S.; Rebizant, J. Behavior of actinide dioxides under pressure: UO22 and ThO2. Phys. Rev. B 2004, 70, 014113. [Google Scholar] [CrossRef]
- Macedo, P.; Capps, W.; Wachtman Jr, J. Elastic constants of single crystal ThO2 at 25 °C. J. Am. Ceram. Soc. 1964, 47, 651–651. [Google Scholar] [CrossRef]
- Stefanovich, E.; Shluger, A.L.; Catlow, C. Theoretical study of the stabilization of cubic-phase ZrO2 by impurities. Phys. Rev. B 1994, 49, 11560. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.-K.; Fang, Y.; Grimsditch, M.; Li, Z.; Nevitt, M.V.; Robertson, W.M.; Zouboulis, E.S. Temperature Dependence of the Elastic Moduli of Monoclinic Zirconia. J. Am. Ceram. Soc. 1991, 74, 1742–1744. [Google Scholar] [CrossRef]
- Nevitt, M.; Chan, S.-K.; Liu, J.; Grimsditch, M.; Fang, Y. The elastic properties of monoclinic ZrO2. Phys. B+C 1988, 150, 230–233. [Google Scholar] [CrossRef]
- Dalton, J.; Janes, P.; Jones, N.; Nicholson, J.; Hallam, K.; Allen, G. Photocatalytic oxidation of NOx gases using TiO2: a surface spectroscopic approach. Environ. Pollut. 2002, 120, 415–422. [Google Scholar] [CrossRef]
- Liu, X.; Fu, J. Electronic and elastic properties of the tetragonal anatase TiO2 structure from first principle calculation. Optik 2020, 206, 164342. [Google Scholar] [CrossRef]
- Burdett, J.K.; Hughbanks, T.; Miller, G.J.; Richardson Jr, J.W.; Smith, J.V. Structural-electronic relationships in inorganic solids: powder neutron diffraction studies of the rutile and anatase polymorphs of titanium dioxide at 15 and 295 K. J. Am. Chem. Soc. 1987, 109, 3639–3646. [Google Scholar] [CrossRef]
- Arlt, T.; Bermejo, M.; Blanco, M.; Gerward, L.; Jiang, J.; Olsen, J.S.; Recio, J. High-pressure polymorphs of anatase TiO2. Phys. Rev. B 2000, 61, 14414. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | a0 (Å) | Egap (eV) | µB | B0 (GPa) | Ecoh (eV/UO2) |
|---|---|---|---|---|---|
| DFT + U [45] | 5.475 | 2.13 | 2.01 | 213 | - |
| LDA + U [46] | 5.474 | 2.06 | 1.35 | 210 | - |
| GGA + U [47] | 5.52 | 1.8 | 1.94 | 209 | −21.7 |
| GGA + U a | 5.497 | 2.14 | 1.99 | 207.2 | −22.3 |
| Exp. | 5.473 [44] | 2.1 [48] | 1.74 [49] | 209 [50] | −22.0 [51] |
| Materials | Incorporation Energy (eV) | Solution Energy (eV) | |
|---|---|---|---|
| O-Rich | U-Rich | ||
| UO2-Th | −10.25 | −11.76 | −0.83 |
| UO2-Zr | −8.68 | −10.19 | 0.74 |
| UO2-Ti | −5.69 | −7.2 | 3.73 |
| Materials | Centre Atom | Nearest O Atom | Nearest U Atom |
|---|---|---|---|
| UO2 | 2.50 (U) | −1.25 | 2.50 |
| UO2-Th | 2.64 (Th) | −1.30 | 2.51 |
| UO2-Zr | 2.48 (Zr) | −1.31 | 2.57 |
| UO2-Ti | 2.09 (Ti) | −1.28 | 2.57 |
| Materials | ∆ (X-U)/U-U | ∆ (X-O)/U-O | ∆V/V0 |
|---|---|---|---|
| UO2-Th | 0.15% | 0.65% | 0.16% |
| UO2-Zr | −0.79% | −3.85% | −0.52% |
| UO2-Ti | −1.33% | −2.78% | −0.88% |
| Materials | UO2 | UO2-Th | UO2-Zr | UO2-Ti |
|---|---|---|---|---|
| Xe1 | 5.17 | 5.12 | 4.96 | 3.12 |
| Xe2 | 1.96 | 1.60 | 1.47 | 1.17 |
| Xe3 | 3.11 | 2.68 | 2.22 | 0.89 |
| Materials | VInt1 (Å3) | VInt2 (Å3) |
|---|---|---|
| UO2 | 21.26 | 21.26 |
| UO2-Xe1 | 22.47 | 22.47 |
| UO2-Xe2 | - | 26.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, L.; Wang, Z.; Wang, Q.; Liu, Z.; Pan, M.; Huang, Z.; Wu, L. Influence of Th, Zr, and Ti Dopants on Solution Property of Xe in Uranium Dioxide with Defects: A DFT + U Study. Metals 2022, 12, 879. https://doi.org/10.3390/met12050879
Pan L, Wang Z, Wang Q, Liu Z, Pan M, Huang Z, Wu L. Influence of Th, Zr, and Ti Dopants on Solution Property of Xe in Uranium Dioxide with Defects: A DFT + U Study. Metals. 2022; 12(5):879. https://doi.org/10.3390/met12050879
Chicago/Turabian StylePan, Liu, Zhen Wang, Qingqing Wang, Zhixiao Liu, Min Pan, Zheng Huang, and Lu Wu. 2022. "Influence of Th, Zr, and Ti Dopants on Solution Property of Xe in Uranium Dioxide with Defects: A DFT + U Study" Metals 12, no. 5: 879. https://doi.org/10.3390/met12050879
APA StylePan, L., Wang, Z., Wang, Q., Liu, Z., Pan, M., Huang, Z., & Wu, L. (2022). Influence of Th, Zr, and Ti Dopants on Solution Property of Xe in Uranium Dioxide with Defects: A DFT + U Study. Metals, 12(5), 879. https://doi.org/10.3390/met12050879