Abstract
This paper shows the results of applying a combination of high energy processing and magnesium (Mg) as an alloying element in a strategy for enhancing the degradation rate of iron (Fe) for applications in the field of non-permanent medical implants. For this purpose, Fe powder was milled with 5 wt% of Mg (Fe5Mg) and its microstructure and characterized degradation behavior. As-received Fe powder was also milled in order to distinguish between the effects due to high energy processing from those due to the presence of Mg. The powders were prepared by high energy planetary ball milling for 16 h. The results show that the initial crystallite size diminishes from >150 nm to 16 nm for Fe and 46 nm for Fe5Mg. Static degradation tests of loose powder particles were performed in Hanks’ solution. Visual inspection of the immersed powders and the X-ray diffraction (XRD) phase quantification indicate that Fe5Mg exhibited the highest degradation rate followed by milled Fe and as received Fe, in this order. The analysis of degradation products of Fe5Mg showed that they consist on magnesium ferrite and pyroaurite, which are known to present good biocompatibility and low toxicity. Differences in structural features and degradation behaviors of milled Fe and milled Fe5Mg suggest the effective dissolution of Mg in the Fe lattice. Based on the obtained results, it can be said that Fe5Mg powder would be a suitable candidate for non-permanent medical implants with a higher degradation rate than Fe.
1. Introduction
Iron (Fe) and magnesium (Mg) are two of the most promising biodegradable, bioabsorbable, and biocompatible metals for temporary medical implants in vascular intervention and osteosynthesis applications [1,2,3,4,5,6,7,8]. Both elements are essential for living organisms and possess low toxicity.
Fe presents interesting mechanical properties that resemble those of SS316L, often used as coronary stent material [9,10], but has a too-low degradation rate [11]. In order to obtain more suitable properties, different candidate alloying elements have been tested. Among them, Mn is the most investigated [2]. This element improves corrosion behavior, increases strength and elongation, and reduces magnetic susceptibility. Other elements that may confer some advantages are C, Si, Co, and Pd [4]. Powder metallurgical processing also may present some benefits, as it permits the combination of different elements and microstructures [12,13]. Electroforming technology is also able to improve Fe performance through the fine grain microstructure that it confers to the material [14].
Another approach to develop biodegradable implant materials is the use of Mg as a base metal [8,15], which presents several distinct properties, among them, excellent biocompatibility, Young’s modulus closer to that of human bones, which prevents stress yielding problems, and antibacterial properties [16,17,18,19]. However, the degradation rate of commercial purity Mg is far too fast. Several strategies have been successful in tailoring its corrosion [20] and degradation rate in order to adjust it to the rate needed for intended applications [6,7,21,22]. These are, among others, the use of high purity Mg and Mg alloys (Mg-Ca, Mg-Zn, and Mg-Ca-Zn) [23], the addition of rare earth elements [8] and the refinement and amorphization of the microstructure [24].
The present investigation starts from the hypothesis that a combination of both elements in solid solution in a Fe-rich range of the phase diagram may have suitable properties as material for biodegradable medical applications, with some advantages when compared with the bare metallic Fe, i.e., faster degradation rate [4,25].
The critical point in the Fe-Mg system is that Fe and Mg are immiscible in solid state in the whole composition range. This represents a challenge when trying to synthesize a binary Fe-Mg solid solution, which requires the use of out-of-equilibrium processing techniques. Early attempts to prepare Fe-Mg were performed by alternate deposition [26] and co-evaporation [27] in ultrahigh vacuum, where thin films were obtained. Processing by high energy ball milling was faced in 1997 by Hightower et al. [28], having in mind hydrogen storage applications. They were able to produce single-phase bcc Fe(Mg) alloys with a concentration of up to about 20 at% (9.8 wt%) Mg. In 2004 and 2005, Dorofeev et al. [29] and Yelsukov et al. [30] presented a study on the kinetics of mechanical alloying and miscibility of this system. They found that the solid solution of Mg in Fe varied non-monotonically in a given range of Mg content. In 2007, Berziou et al. [31] explored the system Fe(1 − x)Mgx, x ≤ 43.4 at% (25 wt%), as a candidate to act as sacrificial coating in the protection of steel structures. The processing method employed was the deposition of Mg by dc magnetron sputtering by which they obtained a solid solution of Mg in Fe, of up to 25 at% (12.65 wt%) of Mg, whereas above this amount amorphization occurred.
The first time that Fe-Mg alloys were proposed for medical degradable applications was in a patent by Guangyin and Jialin [32] released in 2012, where it was claimed that powder metallurgical Fe-xMg alloys (x = 0.1–10 wt%) obtained by ball milling and consolidated by spark plasma sintering improves the degradation rate of the Fe matrix. Research that found an acceleration effect of Mg in combination with Fe employed a mixture of powders in a proportion of Fe to Mg of 99.5 to 0.5 [33]. A similar acceleration effect was obtained in a TCP composite loaded with 30 vol% of cold-sintered Fe-xMg (x = 30 or 50 at%) [34]. Xie et al. [35] compared the degradation rate of a consolidated ball milled Fe-30Mg at% (15.7 wt%) alloy with that of pure Mg and concluded that their Fe-Mg alloy degraded slower. More recently, Shuai et al. [36] also found an acceleration effect of Mg on the degradation rate of an Fe matrix at a specific Mg concentration. The main objective of the present research is to process and characterize an Fe-5 wt% Mg alloy and Fe powders in order to investigate their degradation behavior in Hanks’ pseudophysiological medium and separate the effects caused by high energy ball milling from those caused by the addition of Mg in a metastable solid solution. Special emphasis was put on determining the nature of the degradation products, which, to the authors’ knowledge, is the first time they are reported.
The development of biocompatible absorbable Fe-Mg alloys with a faster degradation rate than Fe might enable the use of this material in a variety of applications, such as graft powder, reinforcing particles of biodegradable polymers, bulk material after consolidation, and feedstock for the additive manufacture of implants.
2. Materials and Methods
Spherical commercial Fe powder particles (99.7% purity, diameter ≤ 74 µm) and spherical Mg powder particles (99.8% purity, diameter ≤ 100 µm) were used in this investigation. For the Fe/Mg ratio, a proportion of 95 wt% Fe to 5 wt% Mg was selected, and the alloy will be referred to as Fe5Mg.
High energy ball milling of the Fe5Mg and Fe powders was carried out in a PM-4 Retsch planetary mill (Retsch GmbH, Haan, Germany) at 200 rpm for up to 16 h and the milled powders, named Fe5Mg16h and Fe16h. The ball (stainless steel, 20 mm diameter) to powder ratio was 7:1. In order to minimize oxidation during the milling process, the powders were introduced in stainless steel mill jars and sealed inside a glove box filled with argon. In addition to the Fe5Mg16h and Fe16h powders, a set of pure Mg was also ball milled following the same procedure and designated as Mg16h. As-received Fe powder was employed as reference materials. The selection of the Fe5Mg composition was based on a preliminary study performed by the authors that suggested that this alloy presented the highest level of Mg in solid solution in the bcc Fe lattice given the absence of Mg peaks in the X-ray diffraction patterns and the highest interplanar distance compared with the other tested compositions. These results are shown in Tables S1 and S2 in the Supplementary Material.
Microstructural characterization was performed by scanning electron microscopy (SEM) (Hitachi High-Tech Corporation, Tokyo, Japan) with a HITACHI S4800 microscope equipped with energy dispersive X-ray spectroscopy (EDS). To observe the cross sections, powders were cold mounted to avoid any phase transformation during the metallographic preparation. Final polishing was achieved with colloidal silica.
Identification of phases and determination of their weight fractions were performed by X-ray diffraction (XRD) with a Bruker AXS D8 diffractometer (Bruker, Billerica, MA, USA) equipped with a Co X-ray tube working at a current of 30 mA and a voltage of 40 kV. The XRD data were collected over a 2θ range of 30–120° with a step size of 0.02°. Quantitative Rietveld analysis was performed and the phase fraction, crystallite size, and lattice parameters were calculated. It is worth mentioning that the Co X-ray tube is especially suited for iron-rich samples as it avoids interferences due to florescence, which do appear with the Cu X-ray tube, precluding the detection of Fe oxides.
Degradation behavior of the Fe5Mg16h, Fe16h, and Fe powders was studied in vitro by immersion of the powders in modified Hanks’ solution (pH 7.4) at 37 °C for 3 and 14 days [37]. The powders with the solution in a ratio of 1 g/50 mL were put in sealed polypropylene test tubes to avoid evaporation. After each immersion time, the powders were centrifuged v = 6000 min−1 (Rotofix 32 Hettich) and the solid rich suspension deposited at the bottom of the FalconTM tubes was mixed with 70% ethanol solution, vortexed, and centrifuged again. This procedure was repeated twice before drying in a fume hood at room temperature. The pH evolution was measured every two days with a pH meter (Checker Plus, Hanna, Hanna Instruments, Woonsocket, RI, USA) with an error of ±0.05.
The description of the milling process of the powders and the immersion degradation tests can be followed in the scheme provided in Figure 1.
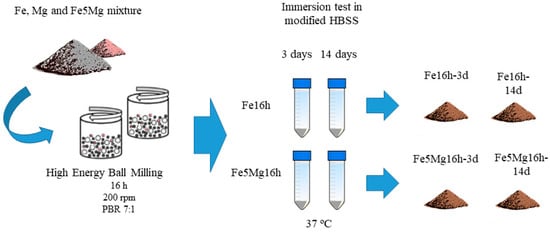
Figure 1.
Scheme of the milling process and degradation test.
3. Results
3.1. Characterization of As-Milled Powders
Figure 2 shows the cross section of Fe5Mg powder after 16 h of ball milling. The layered structure in Figure 2, that was also observed in Fe16h, is typical of a fracturing and cold-welding mechanism that further fractured due to work-hardening. Semiquantitative elemental analysis of a large area of the Fe5Mg16h powder showed Mg contents in accordance with the nominal composition, indicating that no material was preferentially lost during processing.

Figure 2.
Cross sections of Fe5Mg powder ball milled for 16 h. Backscattered scanning electron micrographs at low (a), and high (b) magnifications.
The milled Fe5Mg16h and Fe16h powders were analyzed by the Rietveld refinement of X-ray diffraction data. Figure 3 shows the XRD patterns of both milled powders together with the as-received Fe, Mg16h, and the as-received Mg. By comparing the milled and non-milled unalloyed powders, it is evident that the high energy process caused a broadening of the peaks. This is a typical effect of milling, which produces a reduction in crystallite size and an increase in strain [38]. Table 1 lists the interplanar distances and crystallite sizes. There is no clear difference between interplanar distances of non-milled and milled powders both for Fe and Mg. With respect to the crystallite size, ball milling reduces it significantly, from >150 nm in the as-received powders to 15 nm (α-Fe) in the milled Fe16h powder, and 46 nm (Mg) in the milled Mg16h powder, which confirms the capacity of high-energy processing in the formation of nanocrystallites. On the other hand, the Fe5Mg16h powder presents a four-time larger α-Fe crystallite size, 69 nm against 15 nm, which indicates that the presence of Mg restricts a further reduction of the alloy crystallite size.
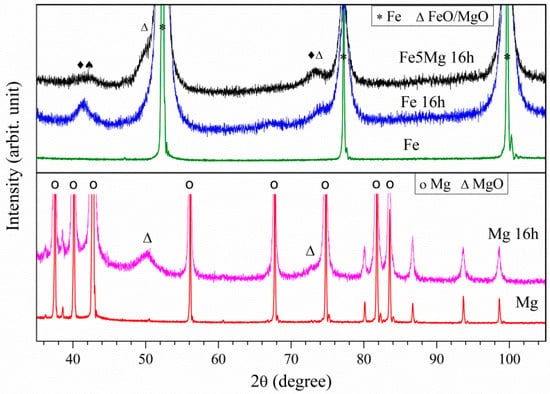
Figure 3.
XRD of as-processed Fe5Mg16h, Fe16h, Fe, Mg16h, and Mg (arbitrary units).

Table 1.
Interplanar distances (ID, nm) corresponding to the [110] planes of α-Fe; and [] and [0001] planes of Mg; and crystallite sizes (CS, nm).
After milling, some phases change in proportion and new ones appear. Phases and phase percentages, calculated from the X-ray diffraction patterns of the powders through Rietveld analysis program TOPAS (Bruker, Billerica, MA, USA), are shown in Table 2. By applying mass balance equations, it is possible to determine the amount of Fe or Mg that reacted during milling. In the as-received Fe powder, 16-h milling gave rise to the formation of 6 wt% magnetite (Fe3O4). If only Fe atoms are considered, it results that the 6 wt% Fe3O4 of the milled powder contains 4.4 wt% of the total amount of Fe, which is the amount that reacted during milling to form part of the oxide phases, whereas 95.6% of the initial Fe remained as zero-valent Fe.
Table 2.
Proportion of phases (wt%) resulting from Rietveld analysis of as-processed and degraded Fe, Fe16h, and Fe5Mg16h powders for 3 and 14 days.
A similar calculation can be performed with the Mg powder to determine the amount of this element that oxidized during the milling process. In this case, the as-received powder was already slightly oxidized as MgO in an amount of 1.2 wt%. This 1.2 wt% MgO contains 0.7 wt% of the total Mg of the powder. After milling for 16 h (Mg16h), the amount of MgO increased up to 12 wt%, which is equivalent to say that the amount of oxidized Mg increased from 0.7 to 7.6 wt%, leaving 92.4 wt% of Mg unoxidized.
In the Fe5Mg16h powder, two new phases formed: 1 wt% of MgFe2O4 (magnesioferrite spinel), and 6 wt% of MgO/FeO (periclase/wüstite). Given the uncertainty in the amount of MgO and FeO, only ranges of Fe and Mg that oxidized as a result of milling can be established. Taking into account the amount of Fe and Mg oxidized during the milling of monolithic powders, the ranges for oxidized Fe and Mg would be from 4.4 to 4.9 wt% and from 13.4 to 7.6 wt%, respectively. By considering these limits, it is possible to calculate that, from the 5 wt% of Mg of the initial powder, between 2.7 and 4.8 wt% is included in a solid solution in α-Fe and the remaining 2.3 to 0.2 wt% reacted during milling and formed part of the two oxides (MgFe2O4 and MgO).
3.2. Characterization of Degraded Powders
The degradation study of Fe5Mg16h, Fe16h, and as-received Fe powders was performed by introducing a given amount of loose powder in Hanks’ solution up to 14 days. A set of powders was cleansed, dried, and studied after 3 days of degradation, and another set after 14 days, when the experiment was finished. For the Fe5Mg16h powder, the reaction with the media started almost immediately and the solution acquired a brownish color after about 2 h of immersion, indicating the formation of oxides. As time increased, in addition to the turbidity of the media, the Fe5Mg16h sample showed an increasing amount of brownish powder. With regard to the pH, it reached a value of 8.6 ± 0.1 after 3 days of immersion and remained invariant afterwards, indicating that the buffering capacity of the media was overwhelmed. A similar, although slower, process was observed in the beaker that contained the milled Fe16h powder. In this case, its pH increased up to 7.7 ± 0.1. In contrast, the as-received Fe powder did not suffer any visible change during the whole test and the pH of its solution remained at 7.4 ± 0.05. These results permit to draw a first conclusion about the sequence of the degradation rate of the studied powders from fast to slow as follows: Fe5Mg16h > Fe16h > Fe.
Following immersion tests, the degraded powders were cleaned, dried, and analyzed by SEM and XRD. Figure 4a,b shows two micrographs of the cross section of the as-received Fe powder that was submerged in Hanks’ solution for 14 days (Fe-14d). At high magnification, a thin grey layer seems to be present around each particle. The corresponding EDS analysis performed in the middle of the powder particles (not shown) presented only a signal of Fe, whereas at their perimeters, some oxygen was detected. However, it was not possible to distinguish whether this oxygen came from the particle surface or from the resin of the mold.

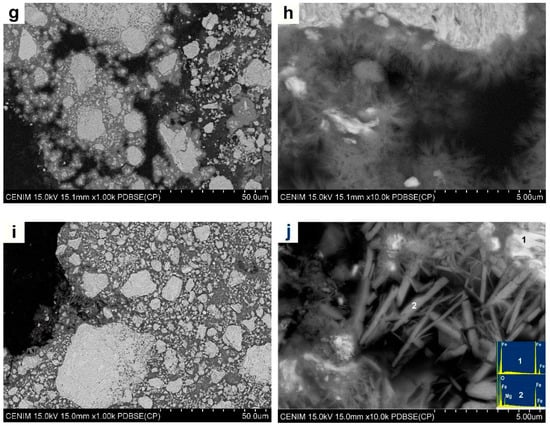
Figure 4.
SEM micrographs of (a,b) as-received Fe powder degraded for 14 days; (c,d) SEM micrographs of Fe16h powder degraded for 3 days; (e,f) Fe16h powder degraded for 14 days; (g,h) Fe5Mg16h powder degraded for 3 days; and (i,j) Fe5Mg16h powder degraded for 14 days.
XRD patterns of the degraded powders are presented in Figure 5 and the obtained phases listed in Table 2. Rietveld analysis of the XRD pattern of Fe-14d indicated that, effectively, there is a small amount of oxide (0.2 wt%) in the form of magnetite (Fe3O4), which was not detected in the non-degraded powder (see Table 2). Given the slow degradation reaction of the as-received Fe powder, no study was performed at 3 days of immersion.
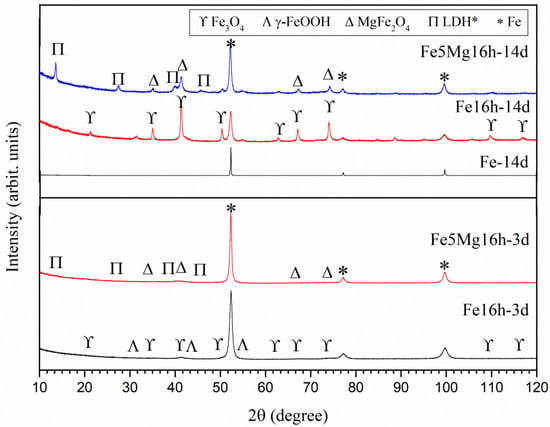
Figure 5.
XRD of the degraded powders after 3 days (Fe5Mg16h and Fe16h-3d), and 14 days (Fe5Mg16h-14d, Fe16h-14d, and Fe-14d) of immersion in Hanks’ solution. Arbitrary units. LDH*: Mg0.75Fe0.25 [OH]2[CO3]0.125·0.5[H2O].
Figure 4c,d shows SEM images of the Fe16h powder after immersion for 3 days in Hanks’ solution (Fe16h-3d). A bright and grey phase can be distinguished. EDS analysis on these zones (not shown) indicated that the bright phase contains only Fe and that the darker one contains Fe and O. Rietveld analysis, Table 2 and Figure 5, shows the presence of 6 wt% of magnetite, almost the same amount as in the as-milled powder, indicating that after 3 days in Hanks’ medium only limited degradation occurred.
Increasing immersion time of the Fe16h powder up to 14 days, Figure 4e,f, resulted in the formation of a large amount of oxides (Table 2 and Figure 5), i.e., 55 wt% of magnetite and 6 wt% of lepidocrocite (γ-FeO(OH)), the latter not detected in the other Fe powders. In addition to the bright α-ferrite, SEM images revealed the presence of a phase that surrounded some granules, and particles that are independent of the granules, all of them with a similar grey contrast, which makes it difficult to distinguish between both reaction phases. From these results, it is evident that ball milling promoted a significant increase in the degradation rate of Fe powder, so that after 14 days of immersion, less than 40 wt% of Fe remained α-ferrite.
The strong influence of the milling process on the degradation behavior of Fe powder can be rationalized by taking into account that milling introduces a high density of crystal defects [38] and promotes a reduction in grain/crystallite size (Table 1) and grain boundaries. These defects act as easy paths for ion diffusion of the degradation medium, which results in an accelerated degradation rate.
Figure 4g,h shows SEM images of the Fe5Mg16h powder degraded for 3 days, where different grey contrasts are observed. A closer examination revealed the presence of a needle-like phase about 1 µm long around the milled Fe5Mg granules, see Figure 4h. As listed in Table 2 and as shown in Figure 5, XRD indicates the presence of 88 wt% α-Fe, 7 wt% of MgFe2O4, and 5 wt% of Mg0.75Fe0.25 [OH]2[CO3]0.125·0.5[H2O] (pyroaurite).
Finally, Figure 4i,j shows micrographs of the Fe5Mg16h powder degraded for 14 days. As occurred with the Fe16h-14d powder, there is a noticeable increase of the greyish (oxide) phases and a decrease of the bright one (α-ferrite) when time of immersion in Hanks’ solution increases from 3 to 14 days. The needle-like phase observed after three days of degradation suffered an increase in size and volume fraction, presenting a length of around 4 µm and a width of about 500 nm. According to XRD analysis, in this material, α-ferrite accounts only for less than 40% of the weight of the degraded powder, being the remaining powder composed by 35 wt% of MgFe2O4 and 35wt% of pyroaurite. Although the amount of α-Fe is similar to that in Fe16h-14d, its result evident from the rapidly change in color of the solution that, during the first stages of immersion, the Fe5Mg16h alloy degraded faster than Fe16h. Some traces of P, Na, and Cl were also found when the samples were analyzed by EDS, but XRD did not show any crystalline phase related to them. The only evidence of a reaction that involves species present in the Hanks’ solution appeared in the XRD pattern as an amorphous background that may be associated to ferrous phosphate precipitates [39], see Figure 5.
4. Discussion
4.1. As-Milled Powders
The goal pursued in this work was to process and characterize an Fe-Mg alloy with a higher degradation rate than pure Fe. In order to get a better insight into the possible degradation mechanisms, pure Fe and Mg were also processed.
After 16 h of milling, Fe16h crystallites reduced their size from >150 to 15 nm, whereas Mg16h crystallites change from >150 to 46 nm. In the case of Mg, this is a saturation value, similar to that encountered by Hwang at al. [40], and Grosjean et al. [41], who employed analogous processing routes, and attributed this behavior to the high recovery rate of magnesium.
The powder formed by Fe and Mg particles is a ductile-brittle couple [38]. Thus, in the initial stages of milling, the Fe ductile metal particles get flattened and cold welded, and the Mg brittle particles get fragmented and trapped in the ductile particles. After enough milling, it is expected that the dispersion of the brittle particles become uniform or enter in solid solution in the Fe matrix, leading to chemical homogeneity. In the Fe5Mg16h powder (Table 1), the crystallite size of α-Fe resulted to 69 nm, more than four times larger than in Fe16h (15 nm), indicating a clear effect of the presence of Mg atoms. Shuai et al. [36] recently prepared Fe-Mg powder (10, 20, 30 at%) by high energy ball milling and obtained grain sizes of 19.3, 12.5, and 11.7 nm, clearly smaller than in the present case, which may be attributed to the different milling parameters (ball to powder ratio of 100:1, rotation speed of 300 rpm, and milling time of 40 h).
Given the argon atmosphere under which the powders were milled, only limited oxidation occurred. For Fe16h, the oxide was Fe3O4 (6 wt%) and for Mg16h, was MgO (12 wt%). In Fe5Mg16h, XRD patterns showed the presence of MgFe2O4 (1 wt%) and MgO/FeO (6 wt%), but no Mg peaks. The absence of Mg signal indicates that some Mg entered in solid solution or the formation of a nanocrystalline structure, as proposed by Swain et al. [34], who prepared biocompatible β-TCP-based composites reinforced with iron–magnesium powder in proportion of about Fe-5Mg and Fe-18Mg (wt%) by high energy milling and did not detect Mg peaks either.
A parameter to evaluate the suitability of the processing route employed in the present work is the amount of Fe and Mg that remained unoxidized after milling, see Table 2. In Fe16h, only 4.4% of Fe appeared in the form of magnetite, which results in 95.6% of unreacted Fe, whereas in the Fe5Mg16h powder, the unreacted Fe was between 94.7 and 95.6. Regarding Mg16h, 7.6% of Mg was associated with MgO, i.e., 92.4% remained unoxidized, whereas in Fe5Mg16h, the unreacted Mg is between 86.3 and 92.4%. The uncertainty in this case is due to the uncertainty in the ratio of MgO/FeO. In all cases, the proportion of metallic Fe and Mg in the milled powders was high and confirms the appropriateness of the processing route. These calculations on the composition of the starting powders aid in the analysis of their in vitro degradation behavior.
4.2. Degraded Powders
Visual inspection of the immersed powders and XRD phase quantification, shown in Table 2, indicate that Fe5Mg16h exhibited the highest degradation rate followed by Fe16h and finally by Fe. It is interesting to note that the as-received Fe powder barely showed any sign of reaction after 14 days of immersion, when only about 0.2 wt% of Fe3O4 was detected. The Pourbaix diagram for the Fe-H2O system predicts that oxidized species will be thermodynamically stable only under certain conditions of potential and pH [42]. More specifically, for the magnetite to form a relatively alkaline, pH is necessary. In consequence, it is reasonable that the small amount of oxidized solid species of Fe detected after 14 days of immersion was associated with the capacity of the Hanks’ solution to buffer an increase of pH, hindering the precipitation of a greater amount of Fe3O4.
In the case of the Fe16h, no significant differences in the amount of magnetite (6 wt%) was detected after 3 days of immersion with respect to that generated during ball milling (6 wt%), and the rest of the solid phase remained as α-Fe. It is known that ball milling introduces microstructural defects during the high energy processing [37]. This is what occurred to the Fe16h powders, as shown in Table 1, where ball milling of the as-received Fe reduced remarkably its crystallite size. Even though this factor is consistent with an increased reactivity and greater amounts of oxidized species after immersion testing, this was not observed. However, this does not mean that ball milling did not enhance the reactivity of the Fe16h powders, during the first three days of immersion. Following the previous rationale, it is reasonable to assume that for the 3-day testing period, the Hanks’ solution buffered the pH conditions to an extent that still prevented magnetite formation. In addition, taking into account that for the as-received Fe, the amount of Fe3O4 was one order of magnitude lower after longer immersion times, it is possible that any increase in the degradation of the Fe16h remained undetected due to the initial amount of magnetite formed mechanically during processing.
In contrast, after 14 days of immersion in Hanks’ solution, only 47 wt% of the milled powder remained as α-Fe and a remarkable increase of magnetite was detected (55 wt%). This is consistent with an increased reactivity leading to enhanced degradation rates. As previously rationalized, this can be explained by a greater number of microstructural defects introduced during high-energy processing. A similar acceleration effect was observed by Bagherifard et al. [43], who promoted grain refinement by severe shot peening of pure iron sheets. Furthermore, the significant amount of Fe3O4 formed is consistent with the fact that the solution pH became more alkaline, favoring oxide precipitation. The degradation of Fe immersed in an aerated electrolyte is an electrochemical process in which the primary cathodic reaction is the reduction of dissolved oxygen, and this reaction results in the formation of hydroxyl ions that locally alkalize the environment. Furthermore, the buffer capacity of a solution depends on the concentration of the weak acid and its conjugate base in the buffer. In the modified Hanks’ solution used in this investigation, the concentration of buffering species such as NaHCO3, HEPES, and HEPES sodium salt were low. Consequently, it is likely that higher degradation rates and longer immersion times contributed to overwhelm the buffer capacity of the test solution, resulting in increasing pH values.
Interestingly, a new phase that was not found previously in the Fe-14d and Fe16h-3d powders was observed. Analysis of the XRD pattern of the Fe16h after 14 days of immersion (Figure 5) determined the presence of lepidocrocite (γ-FeO(OH)) to an extent of about a 6 wt%. It is clear from the evolution of the solid oxides detected by XRD, during immersion of the as-received and the milled Fe powders, that the primary degradation species was Fe3O4, and that the milled condition γ-FeO(OH) appeared only after increasing immersion times. Therefore, even though lepidocrocite may come from the oxidation of zero-valent Fe, it would also be a possibility that γ-FeO(OH) originated from the oxidation of Fe3O4. Unlike Fe3O4, which contains both ferrous (Fe2+) and ferric (Fe3+) ions, in lepidocrocite, all Fe content is fully oxidized. Consequently, the formation of γ-FeO(OH) requires an additional oxidation reaction. As commented above, O2 reduction is the primary cathodic reaction during Fe degradation. In environments where the availability of oxygen is limited, complete oxidation of Fe2+ to Fe3+ is reduced (or even inhibited). In this investigation, the milled powders were immersed in Hanks’ solution inside sealed polypropylene test tubes to avoid evaporation. Therefore, replenishment of the dissolved O2 originally present in the Hanks’ solution that was consumed during Fe oxidation (magnetite formation) was impeded. This limited the amount of Fe3O4 that further oxidized to γ-FeO(OH). Furthermore, lepidocrocite is known to be an oxidant species. Briefly, the ferric ions in γ-FeO(OH) can act as cathodic species and oxidize zero-valent Fe to form Fe2+ ions [44,45], and transform to Fe3O4. For instance, Kassim et al. [46] observed γ-FeO(OH) as a reaction product of iron in an aqueous environment and found that its amount in relation to Fe3O4 depended on the rate of oxidation and the concentration of dissolved oxygen. These factors contribute to explain the remarkable amount of Fe3O4 formed during 14 days of immersion of the Fe16h in comparison with the lower formation of γ-FeO(OH).
In contrast with the Fe and Fe16h powders, the mechanically alloyed Fe-Mg powder prepared in the present work produced Mg-containing phases as degradation products. More specifically, magnesium ferrite (MgFe2O4) and pyroaurite (Mg0.75Fe0.25 [OH]2[CO3]0.125·0.5[H2O]). Compounds of the type pyroaurite are known as layered double hydroxides (LDHs), and they can be generally written as [M(II)1-xM(III)x(OH)2]x+ [(An- x/n)·mH2O]x- in which M(II) and M(III) represent a divalent or trivalent metal cation and An is the anion. LDHs are structures organized in stacked sheets, where the cations reside, while the anion and water move freely in the interlayer region, breaking and forming new bonds [47]. Their crystalline structure can be rhombohedral, like in the case of pyroaurite, or hexagonal (sjögrenite) [48]. Due to their large surface area and their capability of carrying positive charges on the surface, they are used as adsorbents [49]. They have good biocompatibility and low toxicity [50], which make them possible candidates for biomedical applications. On the other hand, MgFe2O4 is a brownish black spinel that belongs to the magnetite series. It is a soft magnetic material used in catalysis, fuel cells, supercapacitor electrodes, microwave devices, etc. [51,52,53]. The nanoparticles are not cytotoxic, present magnetization, and exhibit induction heating. In addition, similar materials [36] showed good cytocompatibility and cell viability. Nevertheless, as some authors expressed [54], there is a concern about the evaluation of Fe-based implants and a thorough analysis is still needed.
Synthesis of MgFe2O4 has been a subject of interest in the recent years. It is produced conventionally by solid state reactions of oxide or carbonate mixtures at elevated temperatures, such as 1100 °C or higher [55,56]. Other production methods such as coprecipitation [57], combustion [58], mechanosynthesis [59], electrospinning [60], and hydrothermal synthesis [61] have also shown to be successful. In the present investigation, an amount of about 1 wt% MgFe2O4 formed from the Fe5Mg after 16 h of high energy milling. This is consistent with previous independent works where polycrystalline MgFe2O4 was prepared by ball milling [62]. Besides, as previously commented either FeO or MgO also formed during milling of the powders.
Table 2 shows that after 3 days of immersion of Fe5Mg16h, similar amounts of MgFe2O4 and MgFe-LDH appeared. All preparation methods of MgFe2O4 commented on the above-required higher temperatures than that used for degradation in the present study (37 °C). However, Xu et al. [63] presented a new method for the preparation of MgFe2O4 by aging a suspension of MgFe-LDH at temperatures below 100 °C. They observed the conversion of the MgFe-LDH to MgFe2O4 at temperatures as low as 40 °C. Furthermore, calculation of the ΔHf° and ΔGf° at 25 °C of MgFe2O4 were determined to be lower than those of the MgFe-LDH, which indicates that MgFe2O4 is more stable than the corresponding MgFe-LDH. In contrast, MgFe-LDHs can be easily prepared by coprecipitation of the two metals at a high pH [64]. It is important to note that alternatively to the oxygen reduction reaction being the main cathodic reaction during Fe degradation, hydrogen evolution (HE) is the primary cathodic reaction during Mg dissolution. This is due to the low corrosion potential normally exhibited by Mg, far below the reversible potential for HE. This reaction produces hydroxyl ions which, in addition to the high chemical activity of Mg, are likely to rapidly alkalize the environment close to the degrading powders. In consequence, it is proposed here that during the immersion of Fe5Mg16h, MgFe-LDH formed first and that the amount of magnesium ferrite found was the sum of that originated mechanically by ball milling (before testing) and by thermostatic conversion of the MgFe-LDH with the time of exposure to the test solution. Finally, when the time of immersion increased to 14 days, greater amounts of both Mg-Fe containing phases were measured.
Interestingly, no trace of the Fe oxidized species found during degradation of Fe and Fe16h were detected. It is reasonable that the dissolved species of Fe that originated due to degradation of the Fe5Mg16h followed the pathway proposed above, contributing to the formation of MgFe-LDH instead of Fe3O4 and γ-FeO(OH).
Other examples of degradation studies of Fe-Mg materials are found in Swain et al. [34], Sikora-Jasinska et al. [39,65,66], and Shuai et al. [36]. Swain et al. [34] studied degradation in Ring’s solution of β-TCP-based composites reinforced with the Fe-Mg powder and observed Fe3O4 and Mg(OH) phases as reaction products. They did not observe any phase that contained both Fe and Mg, which may be due to the annealing at 400 °C that was applied to the samples, which may have induced the segregation of components and even the formation of oxides before the immersion test. Sikora-Jasinska et al. [39,65,66] studied degradation in Hanks’ medium of Fe/Mg2Si composites obtained by ball milling the individual powders followed by consolidation through hot rolling, and found some α-FeO(OH) and FeCO3, but no particles containing Fe and Mg. Finally, Shuai et al. [36] prepared Fe-Mg compacted samples and studied their degradation rate in simulated body fluid (SBF). Although reaction products were not defined, they also observed an increase in degradation of compacted Fe-Mg powder in comparison with Fe. Their maximum degradation rate occurred in the alloy with 20 at% Mg (9.8 wt%), the one that retained, after milling, 8.5 at% Mg (3.9 wt%) in solid solution, slightly less than the 5 wt% in the present case.
Based on the preceding results and discussion it can be stated that the Fe5Mg16h powder obtained in this work, as it degrades faster than Fe powder, seems worth to be considered for further research as bioabsorbable implant material. Possible applications of the powder in the biomedical field would be as grafting material, for hyperthermia treatments, as drug delivery vehicle, as a reinforcing element of biodegradable polymer implants, as bulk material after consolidation, and as feedstock for additive manufacture of porous implants.
There remains a number of issues that deserve deeper investigation, such as its possible antibacterial or osteoinduction effects in comparison with Fe16h, which also proved to degrade faster than the as-received Fe powder, or the employment of different processing methods to produce Fe-Mg alloys in different ways, and the effect of postprocessing to obtain partially fully-consolidated material that might be suitable to be considered for non-permanent medical implants.
5. Conclusions
Fe-5 wt% Mg (Fe5Mg) and Fe powders were processed by high energy planetary ball-milling for 16 h, and characterized as-milled and after immersion in Hanks’ solution for up to 14 days. Special emphasis was put on determining the nature of the degradation products and in comparing the degradation behavior of Fe5Mg16h with that of Fe16h and as-received Fe powder in order to distinguish the effects due to high energy processing from those due to the presence of Mg. The results showed that:
- -
- Due to high energy ball milling, crystallite sizes decrease from >150 nm to 69 nm in Fe5Mg16h and to 15 nm in Fe16h;
- -
- Only limited oxidation occurred during milling, which confirms the appropriateness of the processing route;
- -
- The absence of Mg peaks in XRD patterns of Fe5Mg16h suggests the presence of Mg atoms in solid solution within the Fe lattice;
- -
- Static immersion tests showed that the Fe5Mg milled powder degrades faster than milled Fe, and that both powders degrade much faster than as-received Fe. The distortion that Mg produced in the Fe structure, together with the increase in structural defects introduced by the processing route are proposed as the main mechanisms that promoted this increased degradation rate of the Fe5Mg alloy with respect to Fe;
- -
- The degradation products of milled Fe powder were Fe3O4 and γ-FeO(OH. This can be explained in terms of the absence of O2 during the experiments;
- -
- The degradation products of milled Fe5Mg powder were MgFe2O4 and Mg0.75Fe0.25 [OH]2[CO3]0.125·0.5[H2O];
- -
- The fact that the immersion of Fe5Mg16h in Hanks’ solution generated reaction products that are known to present good biocompatibility and low toxicity, which makes this material a suitable candidate for biomedical applications.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/met12010078/s1. Table S1: Interplanar distance, d (nm), of Fe-xMg powders (x = 1, 3, 5, 7 wt%) milled for 2, 8 and 16 h, corresponding to the [110] planes of Fe, and variation, Δd, with respect to the as-received Fe powder (dFe = 0.28659 Å). Table S2: Weight percent of Mg phase in each alloy for each milling time.
Author Contributions
R.G.E.: Validation, Investigation, Writing—Original Draft, Data curation, Visualization; M.M.: Conceptualization, Methodology, Writing—Original Draft, Supervision; S.F.: Writing—Original Draft; M.L.: Conceptualization, Methodology, Investigation, Writing–Original Draft, Visualization, Supervision, Project administration, Funding acquisition; J.R.: Funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support of the Ministry of Science and Innovation of Spain (MICINN) PID2019-104351GB-C21, PCIN-2017-036 (M-era.Net2016:4128, EU-FEDER), and RTI2018-096391-B-C31, and Comunidad de Madrid ADITIMAT-CM (S2018/NMT-4411) are greatly acknowledged. R. Estrada thanks MICINN for FPI grant PRE2020-092118, and S. Fajardo expresses his gratitude to the State Research Agency (MICINN), the Spanish National Research Council (CSIC), and the European Regional Development Fund (ERDF) for their support under project RYC2019-027006-I (AEI/FEDER/UE).
Data Availability Statement
Mendeley Data, V1, https://doi.org/10.17632/y3c35xrhbb.1 (accessed on 26 December 2021).
Acknowledgments
The authors thank J.A. Jiménez and I. Llorente (XRD), M. Maher and A. Tomás (SEM-EDS), M. Acedo (machining), and students J. Colchero, A. Cardeña, and C. Rodríguez-Castañeda (BM).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Peuster, M.; Wohlsein, P.; Brügmann, M.; Ehlerding, M.; Seidler, K.; Fink, C.; Brauer, H.; Fischer, A.; Hausdorf, G.; Care, I. A novel approach to temporary stenting: Degradable cardiovascular stents produced from corrodible metal—Results 6–18 months after implantation into New Zealand white rabbits. Heart 2001, 1000, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermawan, H.; Dubé, D.; Mantovani, D. Development of Degradable Fe-35Mn Alloy for Biomedical Application. In Advanced Materials Research; Trans Tech Publications Ltd.: Bach, Switzerland, 2006; pp. 107–112. [Google Scholar] [CrossRef]
- Hermawan, H.; Dubé, D.; Mantovani, D. Developments in metallic biodegradable stents. Acta Biomater. 2010, 6, 1693–1697. [Google Scholar] [CrossRef]
- Schinhammer, M.; Hänzi, A.C.; Löffler, J.F.; Uggowitzer, P.J. Design strategy for biodegradable Fe-based alloys for medical applications. Acta Biomater. 2010, 6, 1705–1713. [Google Scholar] [CrossRef]
- Witte, F.; Kaese, V.; Haferkamp, H.; Switzer, E.; Meyer-Lindenberg, A.; Wirth, C.J.; Windhagen, H. In vivo corrosion of four magnesium alloys and the associated bone response. Biomaterials 2005, 26, 3557–3563. [Google Scholar] [CrossRef]
- Witte, F.; Fischer, J.; Nellesen, J.; Crostack, H.A.; Kaese, V.; Pisch, A.; Beckmann, F.; Windhagen, H. In vitro and in vivo corrosion measurements of magnesium alloys. Biomaterials 2006, 27, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.F.F.; Gu, X.N.N.; Witte, F. Biodegradable metals. Mater. Sci. Eng. R Rep. 2014, 77, 1–34. [Google Scholar] [CrossRef]
- Han, H.S.; Loffredo, S.; Jun, I.; Edwards, J.; Kim, Y.C.; Seok, H.K.; Witte, F.; Mantovani, D.; Glyn-Jones, S. Current status and outlook on the clinical translation of biodegradable metals. Mater. Today 2019, 23, 57–71. [Google Scholar] [CrossRef]
- Balcon, R. Recommendations on stent manufacture, implantation and utilization. Eur. Heart J. 1997, 18, 1536–1547. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Yang, C.; Lin, S.; Yu, Q.; Wang, G. Biodegradable stents for coronary artery disease treatment: Recent advances and future perspectives. Mater. Sci. Eng. C 2018, 91, 163–178. [Google Scholar] [CrossRef]
- Venezuela, J.; Dargusch, M.S. Addressing the slow corrosion rate of biodegradable Fe-Mn: Current approaches and future trends. Curr. Opin. Solid State Mater. Sci. 2020, 24, 100822. [Google Scholar] [CrossRef]
- Wegener, B.; Sievers, B.; Utzschneider, S.; Müller, P.; Jansson, V.; Rößler, S.; Nies, B.; Stephani, G.; Kieback, B.; Quadbeck, P. Microstructure, cytotoxicity and corrosion of powder-metallurgical iron alloys for biodegradable bone replacement materials. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2011, 176, 1789–1796. [Google Scholar] [CrossRef]
- Gorejová, R.; Haverová, L.; Oriňaková, R.; Oriňak, A.; Oriňak, M. Recent advancements in Fe-based biodegradable materials for bone repair. J. Mater. Sci. 2019, 54, 1913–1947. [Google Scholar] [CrossRef]
- Moravej, M.; Mantovani, D. Biodegradable metals for cardiovascular stent application: Interests and new opportunities. Int. J. Mol. Sci. 2011, 12, 4250–4270. [Google Scholar] [CrossRef] [Green Version]
- Staiger, M.P.; Pietak, A.M.; Huadmai, J.; Dias, G. Magnesium and its alloys as orthopedic biomaterials: A review. Biomaterials 2006, 27, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.A.; Griffith, R.W.; Shechtman, D.; Evans, R.B.; Conzemius, M.G. In vitro antibacterial properties of magnesium metal against Escherichia coli, Pseudomonas aeruginosa and Staphylococcus aureus. Acta Biomater. 2010, 6, 1869–1877. [Google Scholar] [CrossRef]
- Zeng, J.; Ren, L.; Yuan, Y.; Wang, Y.; Zhao, J.; Zeng, R.; Yang, K.; Mei, X. Short-term effect of magnesium implantation on the osteomyelitis modeled animals induced by Staphylococcus aureus. J. Mater. Sci. Mater. Med. 2013, 24, 2405–2416. [Google Scholar] [CrossRef]
- Rodríguez-Sánchez, J.; Pacha-Olivenza, M.Á.; González-Martín, M.L. Bactericidal effect of magnesium ions over planktonic and sessile Staphylococcus epidermidis and Escherichia coli. Mater. Chem. Phys. 2019, 221, 342–348. [Google Scholar] [CrossRef]
- Pacha-Olivenza, M.A.A.; Galván, J.C.C.; Porro, J.A.A.; Lieblich, M.; Díaz, M.; Angulo, I.; Cordovilla, F.; García-Galván, F.R.R.; Fernández-Calderón, M.C.C.; González-Martín, M.L.L.; et al. Efficacy of laser shock processing of biodegradable Mg and Mg-1Zn alloy on their in vitro corrosion and bacterial response. Surf. Coat. Technol. 2020, 384, 125320. [Google Scholar] [CrossRef]
- Song, G. Control of biodegradation of biocompatable magnesium alloys. Corros. Sci. 2007, 49, 1696–1701. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Z.; Smith, C.; Sankar, J. Recent advances on the development of magnesium alloys for biodegradable implants. Acta Biomater. 2014, 10, 4561–4573. [Google Scholar] [CrossRef]
- Zhao, D.; Witte, F.; Lu, F.; Wang, J.; Li, J.; Qin, L. Current status on clinical applications of magnesium-based orthopaedic implants: A review from clinical translational perspective. Biomaterials 2017, 112, 287–302. [Google Scholar] [CrossRef]
- Cihova, M.; Martinelli, E.; Schmutz, P.; Myrissa, A.; Schäublin, R.; Weinberg, A.M.; Uggowitzer, P.J.; Löffler, J.F. The role of zinc in the biocorrosion behavior of resorbable Mg–Zn–Ca alloys. Acta Biomater. 2019, 100, 398–414. [Google Scholar] [CrossRef]
- Kiani, F.; Wen, C.; Li, Y. Prospects and strategies for magnesium alloys as biodegradable implants from crystalline to bulk metallic glasses and composites—A review. Acta Biomater. 2020, 103, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Multigner, M.; Lieblich, M.; González-Carrasco, J.L.; Rodríguez-Castañeda, C.; Cardeña, A.; de la Presa, P.; Rams, J. Characterisation of planetary ball-milled Fe-Mg powder. In Proceedings of the 11th International Symposium on Biodegradable Metals for Biomedical Applications, Alicante, Spain, 25–30 August 2019. [Google Scholar]
- Kawaguchi, K.; Yamamoto, R.; Hosoito, N.; Shinjo, T.; Takada, T. Magnetic Properties of Fe–Mg Artificial Superstructure Films. J. Phys. Soc. Jpn. 1986, 55, 2375–2383. [Google Scholar] [CrossRef]
- Fnidiki, A.; Eymery, J.E.; Denanot, M.E.; de Mdtal, L.; Pineau, R.; Poitiers, F. Structure and stability of Fe-Mg amorphous alloys prepared by coevaporation. Hyperfine Interact. 1989, 45, 295–300. [Google Scholar] [CrossRef]
- Hightower, A.; Fultz, B.; Bowman, R.C. Mechanical alloying of Fe and Mg. J. Alloys Compd. 1997, 252, 238–244. [Google Scholar] [CrossRef]
- Dorofeev, G.A.; Elsukov, E.P.; Ul’anov, A.L. Mechanical Alloying of Immiscible Elements in the Fe-Mg System. Inorg. Mater. 2004, 40, 690–699. [Google Scholar] [CrossRef]
- Yelsukov, E.P.; Dorofeev, G.A.; Ulyanov, A.L. Mechanism and kinetics of mechanical alloying in an immiscible Fe-Mg system. Czechoslov. J. Phys. 2005, 55, 913–921. [Google Scholar] [CrossRef]
- Berziou, C.; Remy, K.; Billard, A.; Creus, J. Corrosion behavior of dc magnetron sputtered Fe1-xMgx alloy films in 3 wt{%} NaCl solution. Corros. Sci. 2007, 49, 4276–4295. [Google Scholar] [CrossRef]
- Guangyin, Y.; Jialin, N. Medical degradable Fe-Mg Binary Alloy Material and Preparation Method Thereof. Chinese Patent CN103028149B, 27 August 2012. [Google Scholar]
- Oriňáková, R.; Oriňák, A.; Bučková, L.M.; Giretová, M.; Medveckỳ, L.; Labbanczová, E.; Kupková, M.; Hrubovčáková, M.; Koval, K. Iron based degradable foam structures for potential orthopedic applications. Int. J. Electrochem. Sci. 2013, 8, 12451–12465. [Google Scholar]
- Swain, S.K.; Gotman, I.; Unger, R.; Kirkpatrick, C.J.; Gutmanas, E.Y. Microstructure, mechanical characteristics and cell compatibility of $β$-tricalcium phosphate reinforced with biodegradable Fe-Mg metal phase. J. Mech. Behav. Biomed. Mater. 2016, 53, 434–444. [Google Scholar] [CrossRef]
- Xie, G.; Takada, H.; Kanetaka, H. Development of high performance MgFe alloy as potential biodegradable materials. Mater. Sci. Eng. A 2016, 671, 48–53. [Google Scholar] [CrossRef]
- Shuai, C.; He, C.; Qian, G.; Min, A.; Deng, Y.; Yang, W.; Zang, X. Mechanically driving supersaturated Fe–Mg solid solution for bone implant: Preparation, solubility and degradation. Compos. Part B Eng. 2021, 207, 108564. [Google Scholar] [CrossRef]
- Lévesque, J.; Hermawan, H.; Dubé, D.; Mantovani, D. Design of a pseudo-physiological test bench specific to the development of biodegradable metallic biomaterials. Acta Biomater. 2008, 4, 284–295. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Sikora-Jasinska, M.; Chevallier, P.; Turgeon, S.; Paternoster, C.; Mostaed, E.; Vedani, M.; Mantovani, D. Long-term in vitro degradation behavior of Fe and Fe/Mg2Si composites for biodegradable implant applications. RSC Adv. 2018, 8, 9627–9639. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Nishimura, C.; McCormick, P.G. Mechanical milling of magnesium powder. Mater. Sci. Eng. A 2001, 318, 22–33. [Google Scholar] [CrossRef]
- Grosjean, M.H.; Zidoune, M.; Roué, L.; Huot, J.; Schulz, R. Effect of ball milling on the corrosion resistance of magnesium in aqueous media. Electrochim. Acta 2004, 49, 2461–2470. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions; National Association of Corrosion Engineers: Houston, TX, USA, 1984. [Google Scholar]
- Bagherifard, S.; Molla, M.F.; Kajanek, D.; Donnini, R.; Hadzima, B.; Guagliano, M. Accelerated biodegradation and improved mechanical performance of pure iron through surface grain refinement. Acta Biomater. 2019, 98, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Antony, H.; Legrand, L.; Maréchal, L.; Perrin, S.; Dillmann, P.; Chaussé, A. Study of lepidocrocite γ-FeOOH electrochemical reduction in neutral and slightly alkaline solutions at 25 °C. Electrochim. Acta 2005, 51, 745–753. [Google Scholar] [CrossRef]
- Liu, A.; Liu, J.; Pan, B.; Zhang, W.X. Formation of lepidocrocite (γ-FeOOH) from oxidation of nanoscale zero-valent iron (nZVI) in oxygenated water. RSC Adv. 2014, 4, 57377–57382. [Google Scholar] [CrossRef]
- Kassim, J.; Baird, T.; Fryer, J.R. Electron microscope studies of iron corrosion products in water at room temperature. Corros. Sci. 1982, 22, 147–158. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Ingram, L.; Taylor, H.F.W. The crystal structures of sjögrenite and pyroaurite. Mineral. Mag. J. Mineral. Soc. 1967, 36, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Guo, Q.; Liang, M.; Sun, W. Sb(III) and Sb(V) removal from water by a hydroxyl-intercalated, mechanochemically synthesized Mg-Fe-LDH. Appl. Clay Sci. 2020, 196, 105766. [Google Scholar] [CrossRef]
- Chen, J.; Feng, J.; Yan, L.; Li, H.; Xiong, C.; Ma, S. In situ growth process of Mg–Fe layered double hydroxide conversion film on MgCa alloy. J. Magnes. Alloy. 2020, 9, 1019–1027. [Google Scholar] [CrossRef]
- Gong, C.; Bai, Y.J.; Qi, Y.X.; Lun, N.; Feng, J. Preparation of carbon-coated MgFe2O4 with excellent cycling and rate performance. Electrochim. Acta 2013, 90, 119–127. [Google Scholar] [CrossRef]
- Leonés, A.; Lieblich, M.; Benavente, R.; Gonzalez, J.L. Potential Applications of Magnesium-Based Polymeric Nanocomposites Obtained by Electrospinning Technique. Nanomaterials 2020, 10, 1524. [Google Scholar] [CrossRef]
- Israr, M.; Iqbal, J.; Arshad, A.; Gómez-Romero, P.; Benages, R. Multifunctional MgFe2O4/GNPs nanocomposite: Graphene-promoted visible light driven photocatalytic activity and electrochemical performance of MgFe2O4 nanoparticles. Solid State Sci. 2020, 110, 106363. [Google Scholar] [CrossRef]
- Scarcello, E.; Lison, D. Are Fe-based stenting materials biocompatible? A critical review of in vitro and in vivo studies. J. Funct. Biomater. 2020, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkin, A.I.; Drebushchak, V.A. Synthesis and calorimetric investigation of stoichiometric Fe-spinels: MgFe2O4. J. Cryst. Growth 2004, 265, 165–167. [Google Scholar] [CrossRef]
- Ilhan, S.; Izotova, S.G.; Komlev, A.A. Synthesis and characterization of MgFe2O4 nanoparticles prepared by hydrothermal decomposition of co-precipitated magnesium and iron hydroxides. Ceram. Int. 2015, 41, 577–585. [Google Scholar] [CrossRef]
- Chen, Q.; Rondinone, A.J.; Chakoumakos, B.C.; Zhang, Z.J. Synthesis of superparamagnetic MgFe2O4 nanoparticles by coprecipitation. J. Magn. Magn. Mater. 1999, 194, 1–7. [Google Scholar] [CrossRef]
- Huang, Y.; Tang, Y.; Wang, J.; Chen, Q. Synthesis of MgFe2O4 nanocrystallites under mild conditions. Mater. Chem. Phys. 2006, 97, 394–397. [Google Scholar] [CrossRef]
- Šepelák, V.; Bergmann, I.; Menzel, D.; Feldhoff, A.; Heitjans, P.; Litterst, F.J.; Becker, K.D. Magnetization enhancement in nanosized MgFe2O4 prepared by mechanosynthesis. J. Magn. Magn. Mater. 2007, 316, 764–767. [Google Scholar] [CrossRef]
- Maensiri, S.; Sangmanee, M.; Wiengmoon, A. Magnesium ferrite (MgFe2O4) nanostructures fabricated by electrospinning. Nanoscale Res. Lett. 2009, 4, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Joy, P.A.; Khollam, Y.B.; Potdar, H.S.; Deshpande, S.B. Synthesis of nanosized MgFe2O4 powders by microwave hydrothermal method. Mater. Lett. 2004, 58, 1092–1095. [Google Scholar] [CrossRef]
- Šepelák, V.; Schultze, D.; Krumeich, F.; Steinike, U.; Becker, K.D. Mechanically induced cation redistribution in magnesium ferrite and its thermal stability. Solid State Ion. 2001, 141–142, 677–682. [Google Scholar] [CrossRef]
- Xu, Q.; Wei, Y.; Liu, Y.; Ji, X.; Yang, L.; Gu, M. Preparation of Mg/Fe spinel ferrite nanoparticles from Mg/Fe-LDH microcrystallites under mild conditions. Solid State Sci. 2009, 11, 472–478. [Google Scholar] [CrossRef]
- Hansen, H.C.B.; Taylor, R.M. Formation of synthetic analogues of double metal-hydroxy carbonate minerals under controlled pH conditions: I. The synthesis of pyroaurite and reevesite. Clay Miner. 1990, 25, 161–179. [Google Scholar] [CrossRef]
- Sikora-Jasinska, M.; Paternoster, C.; Mostaed, E.; Tolouei, R.; Casati, R.; Vedani, M.; Mantovani, D. Synthesis, mechanical properties and corrosion behavior of powder metallurgy processed Fe/Mg2Si composites for biodegradable implant applications. Mater. Sci. Eng. C 2017, 81, 511–521. [Google Scholar] [CrossRef]
- Sikora-Jasinska, M.; Chevallier, P.; Turgeon, S.; Paternoster, C.; Mostaed, E.; Vedani, M.; Mantovani, D. Understanding the effect of the reinforcement addition on corrosion behavior of Fe/Mg2Si composites for biodegradable implant applications. Mater. Chem. Phys. 2019, 223, 771–778. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).