
Recovering Value from End-of-Life Batteries by Integrating Froth Flotation and Pyrometallurgical Copper-Slag Cleaning

 , , ,
, , ,  and
and
Abstract
:1. Introduction
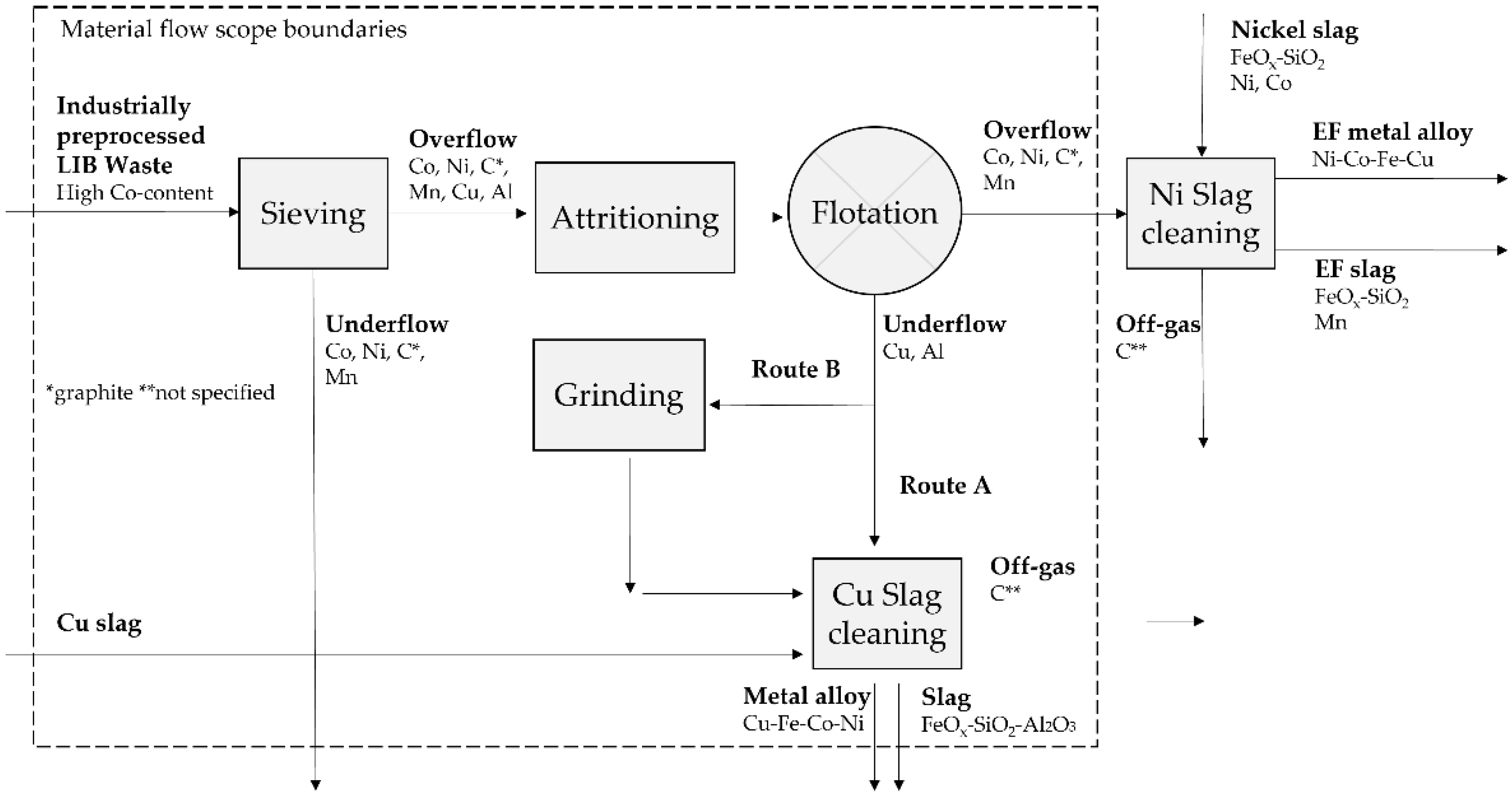
2. Materials and Methods
2.1. Froth Flotation
2.1.1. Sample Preparation
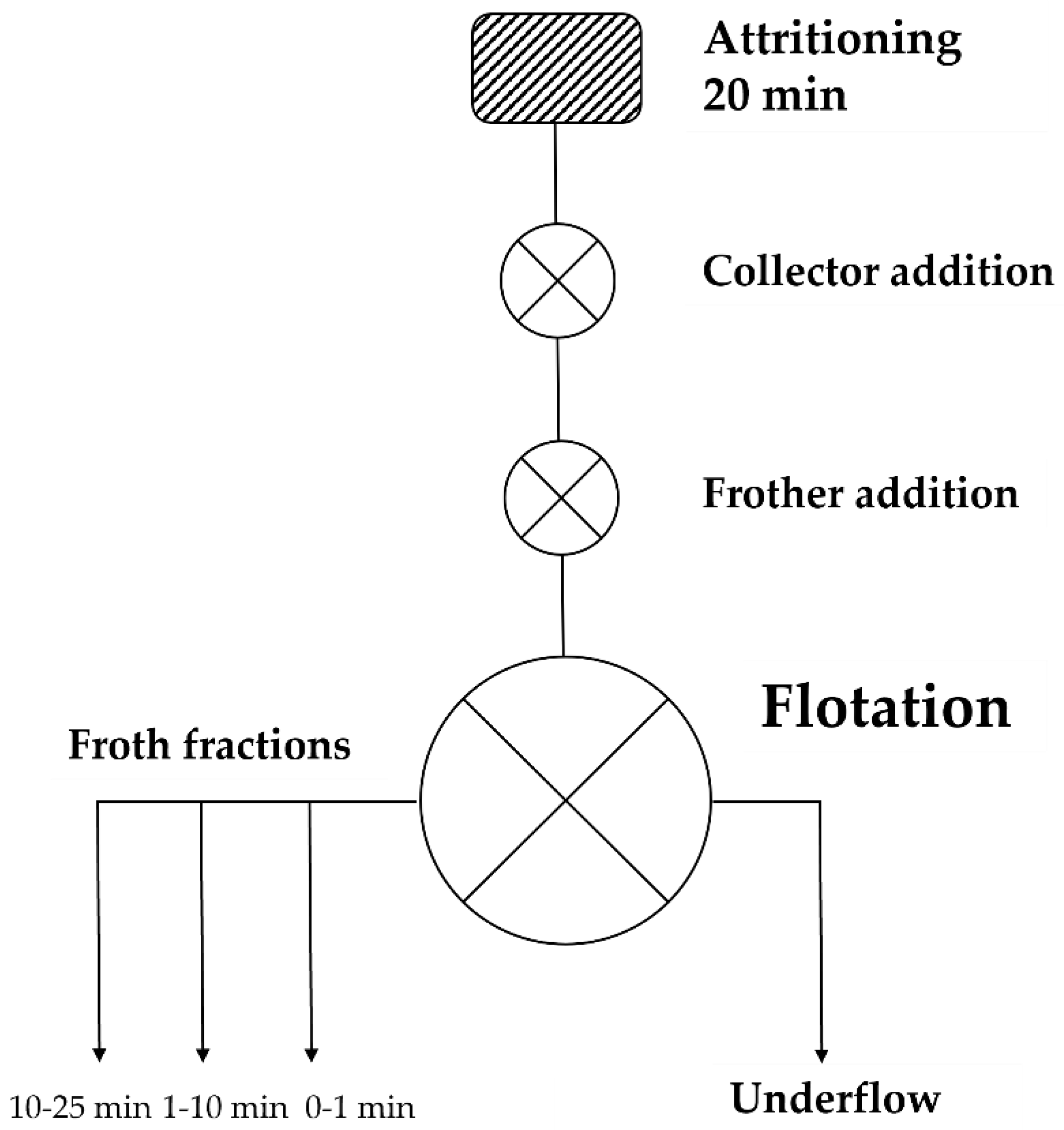
2.1.2. Flotation Experiments
2.1.3. Sample Preparation for Pyrometallurgical Reduction Experiments
2.1.4. Characterization
- C = Fraction of feed weight reporting to the froth/overflow of the sieve (wt.%)
- m = Metal content of Cu (wt.%)
- c = Fraction of Cu reporting to the froth/overflow of the sieve (wt.%)
- f = Fraction of Cu reporting to the feed/underflow of the sieve (wt.%)
2.2. Pyrometallurgy
2.3. Characterization of Sample Composition
3. Results and Discussion
3.1. Froth Flotation Experiments
3.1.1. Chemical Composition of the LIB Waste
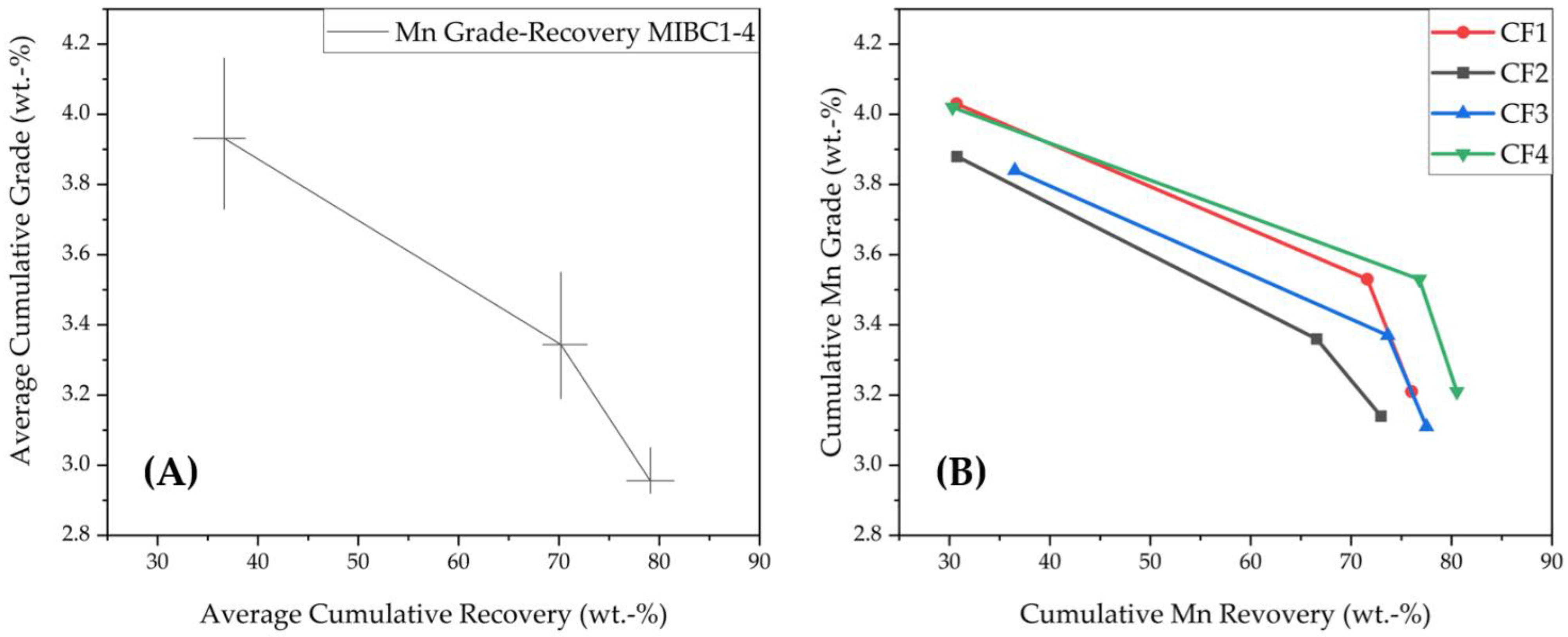
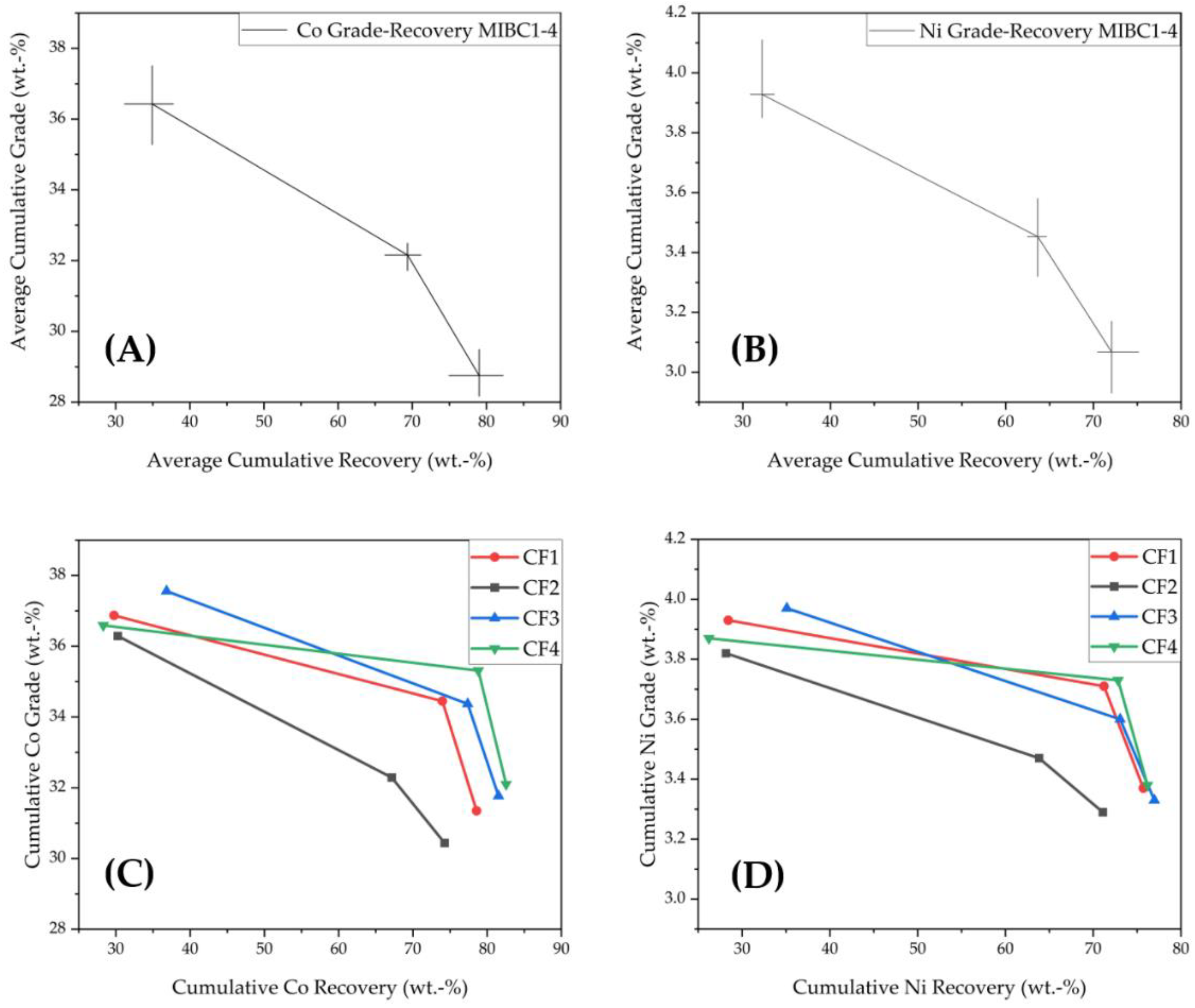
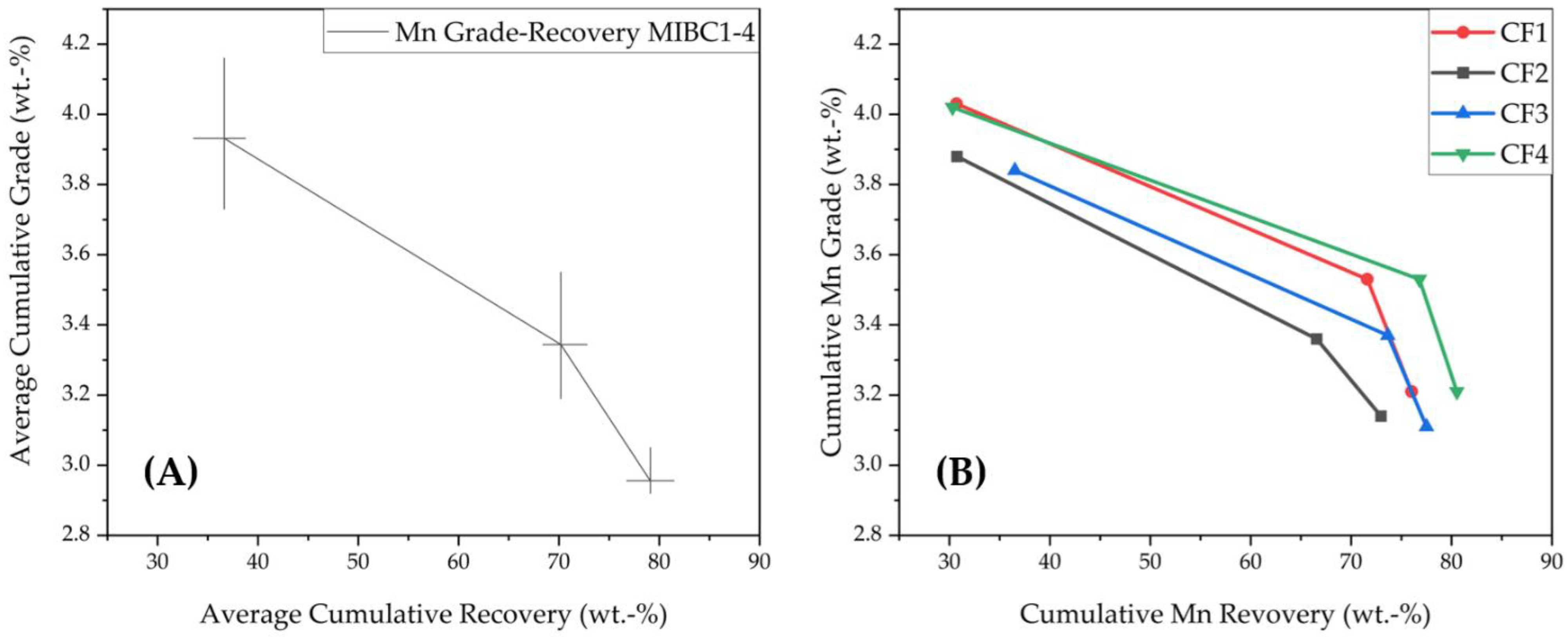
3.1.2. Grade-Recovery Curves for Co and Ni
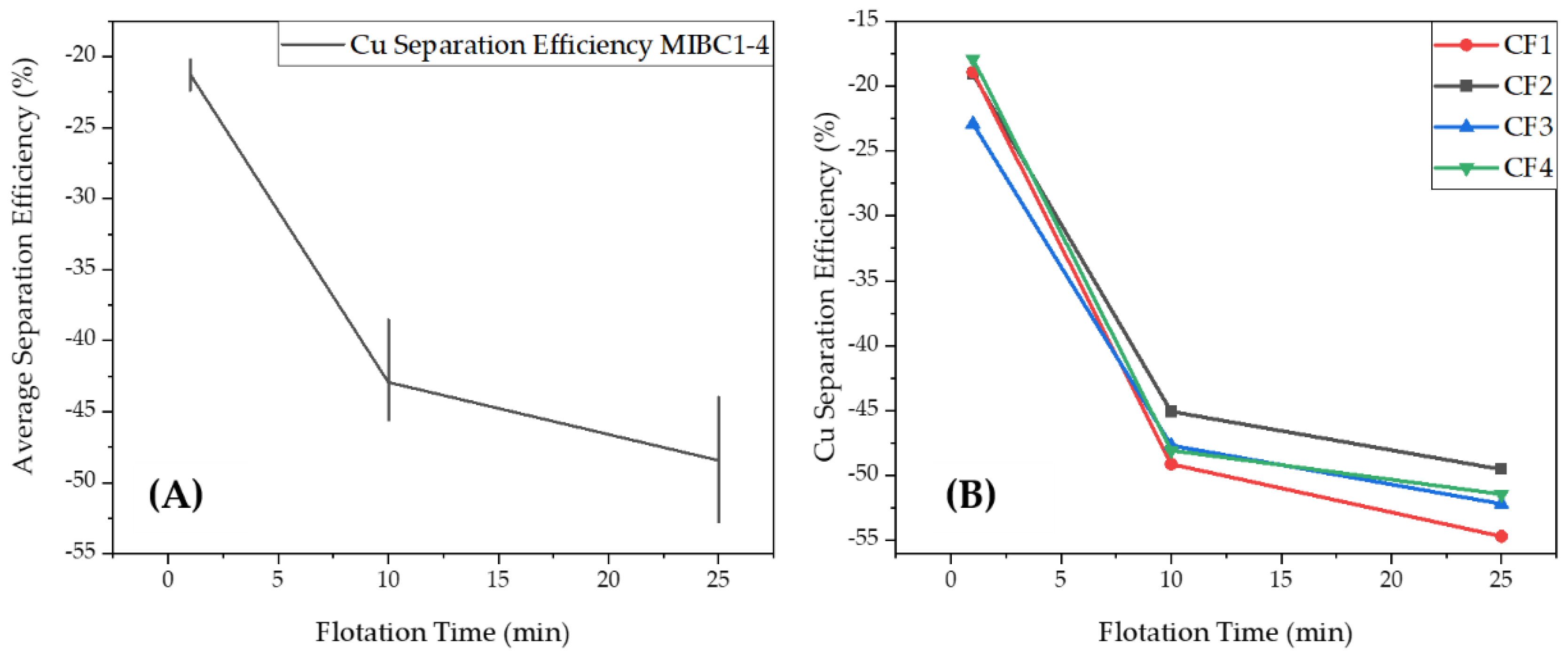
3.1.3. Separation Efficiency of Cu
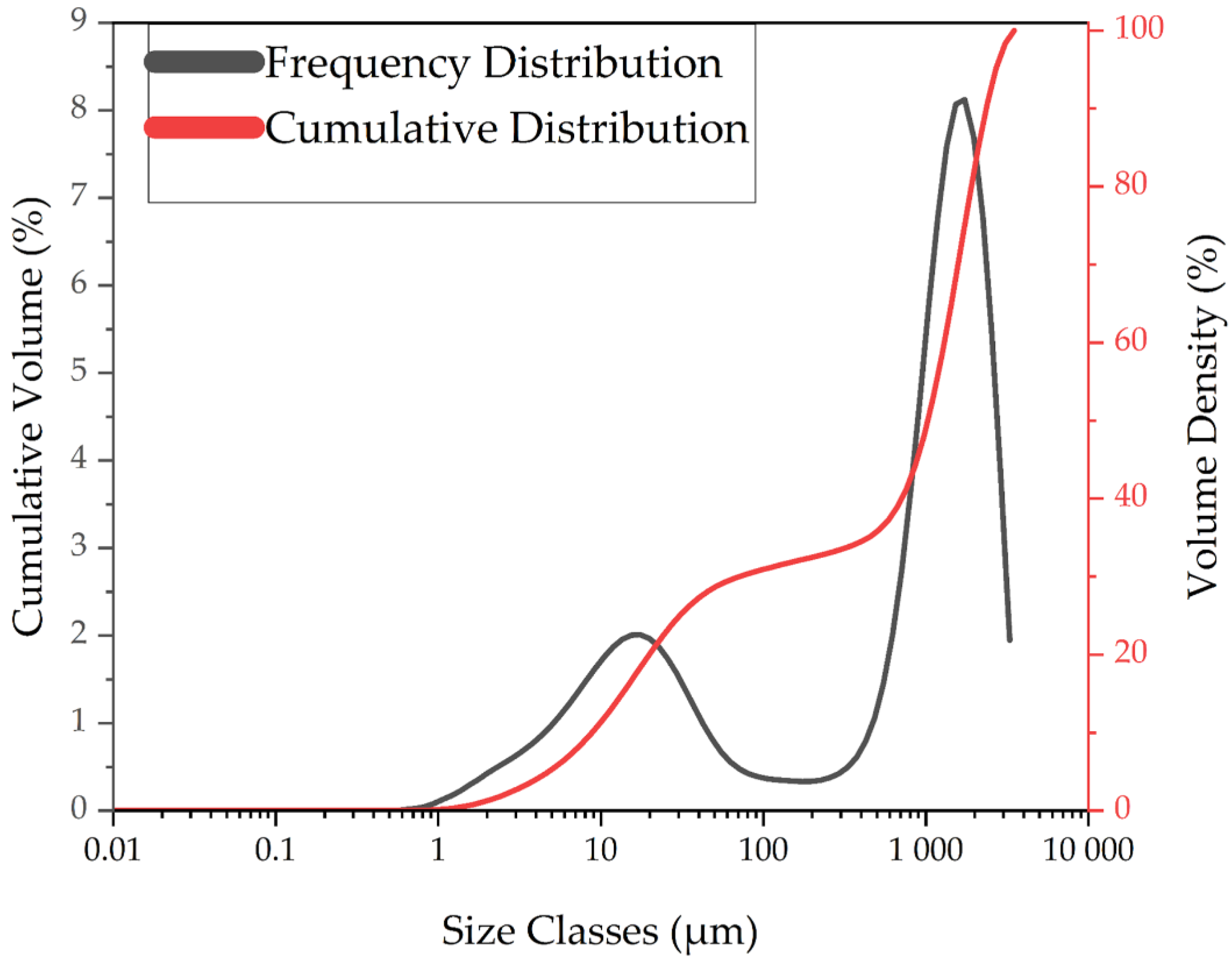
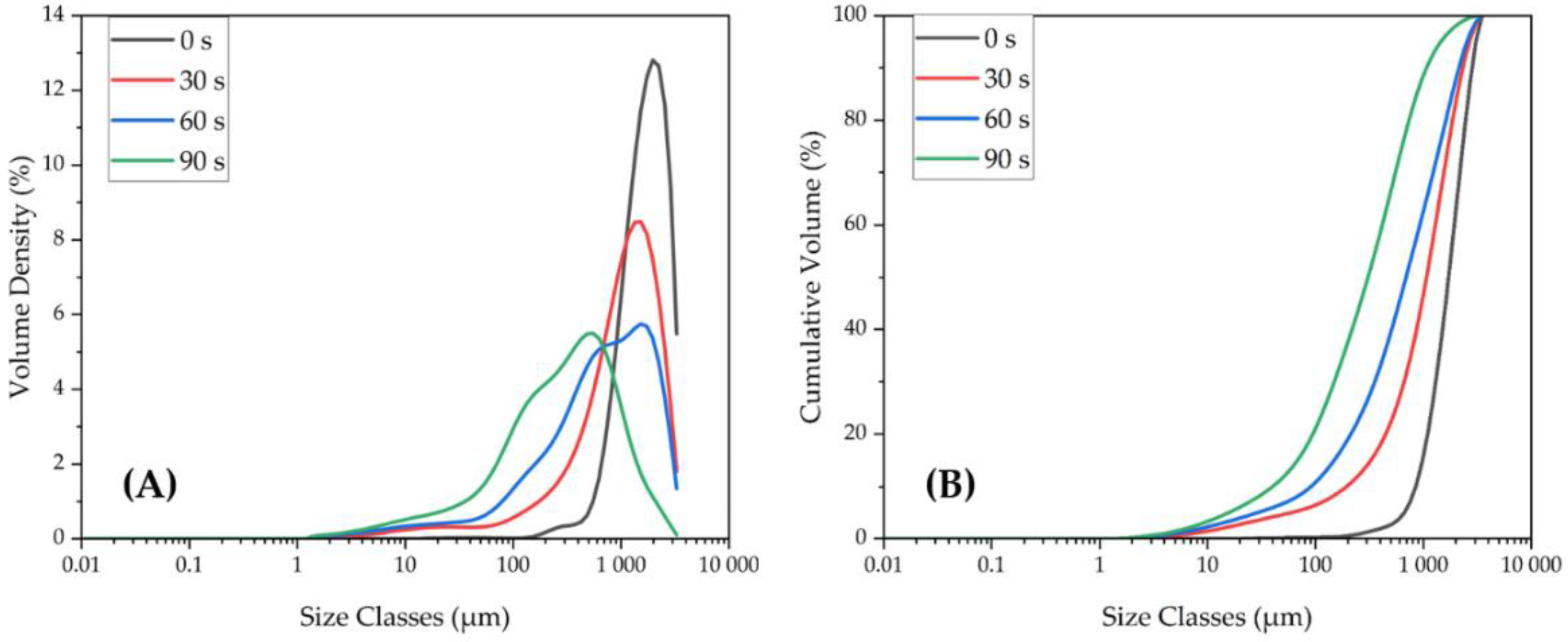
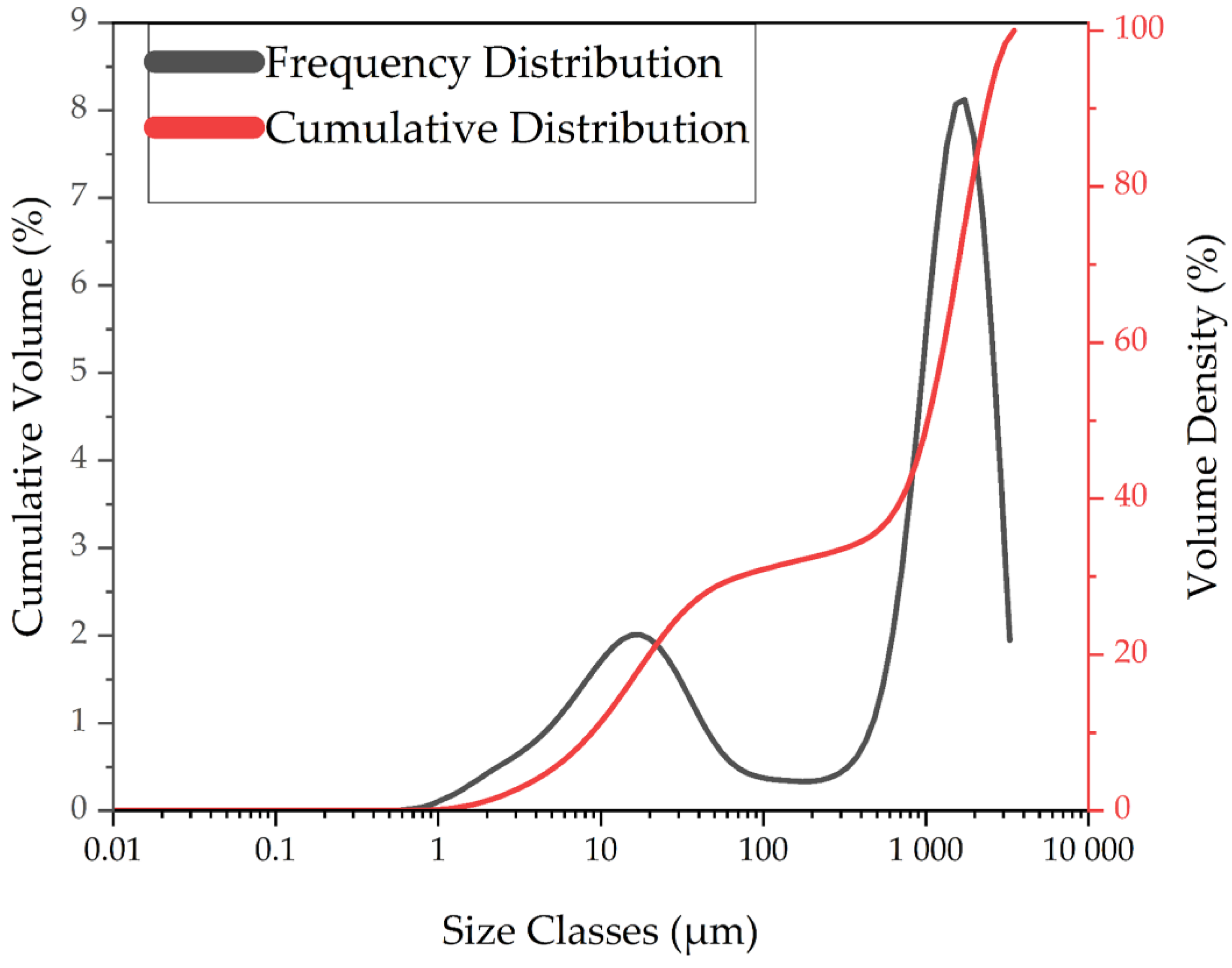
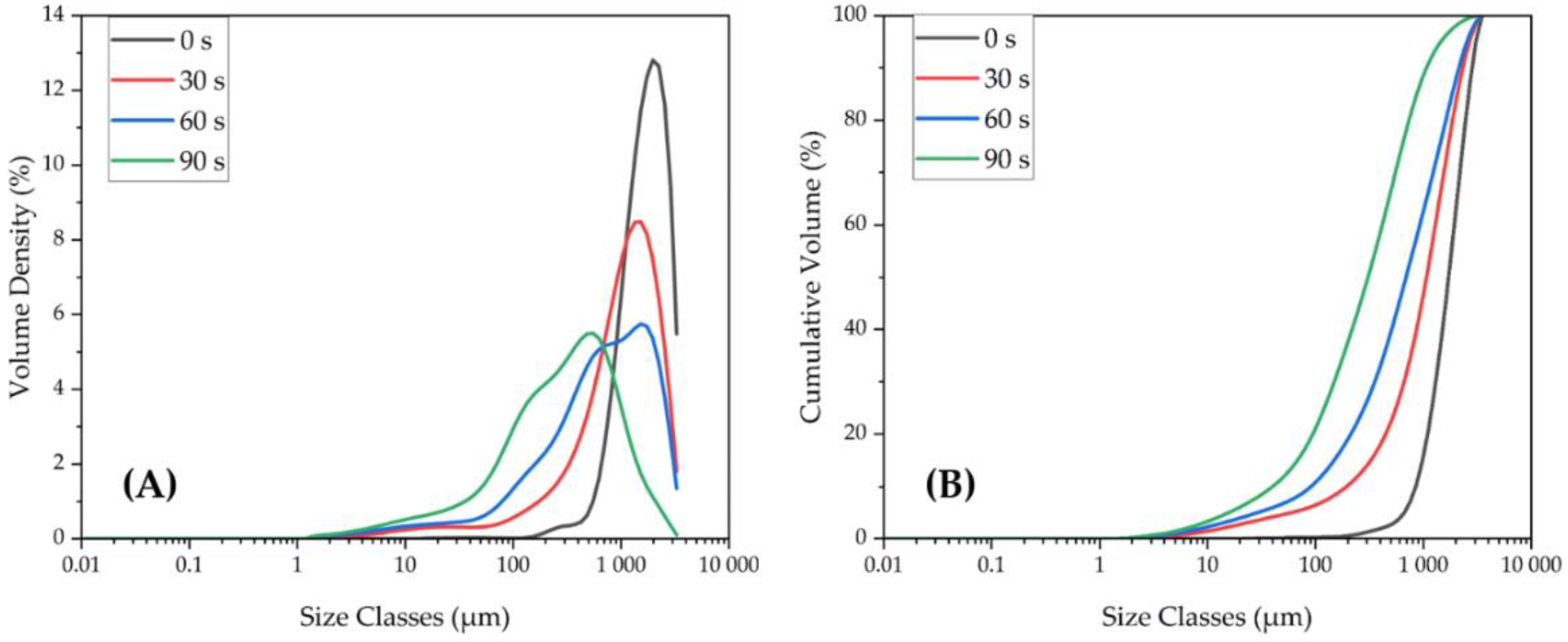
3.1.4. Particle Size Distributions of LIB Waste and Ground Flotation Underflow
3.1.5. Chemical Composition of Flotation Underflow
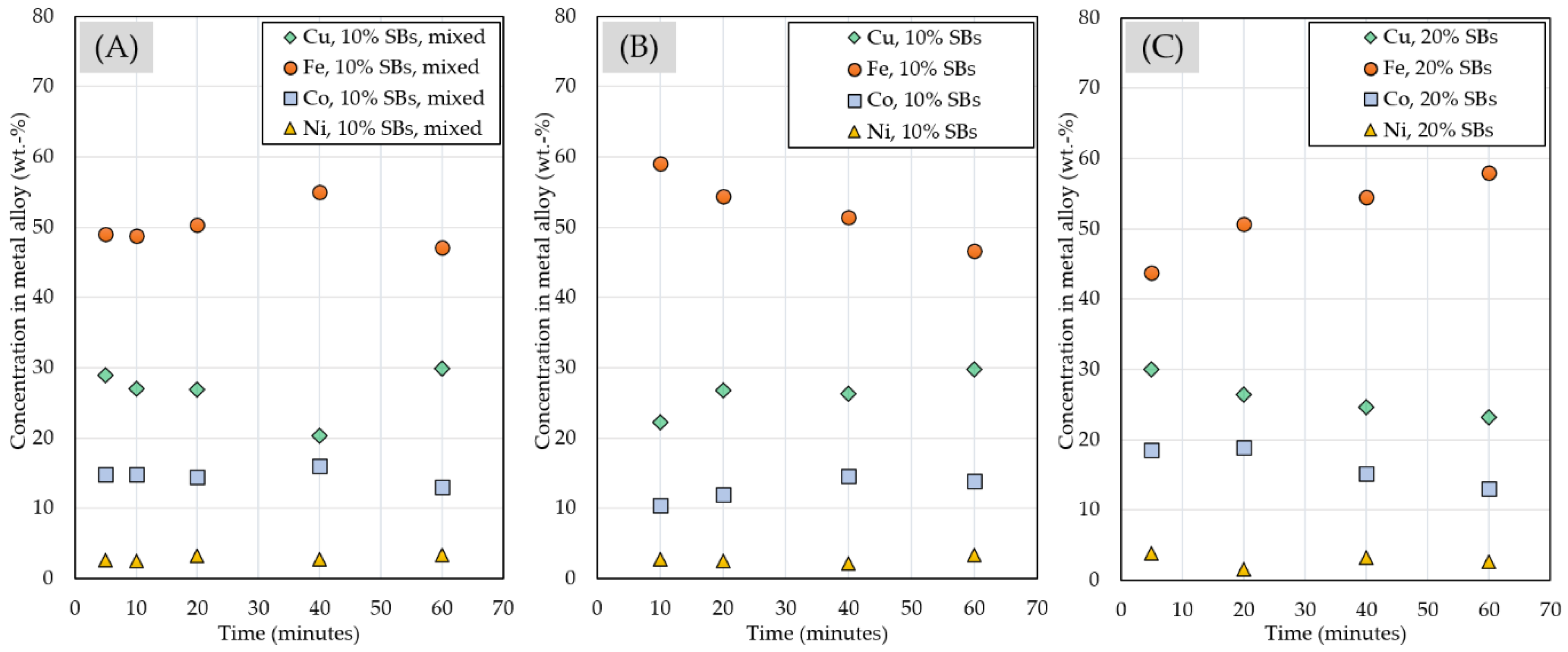
3.2. Pyrometallurgical Treatment
3.2.1. Chemical Composition of the Feed Mixture
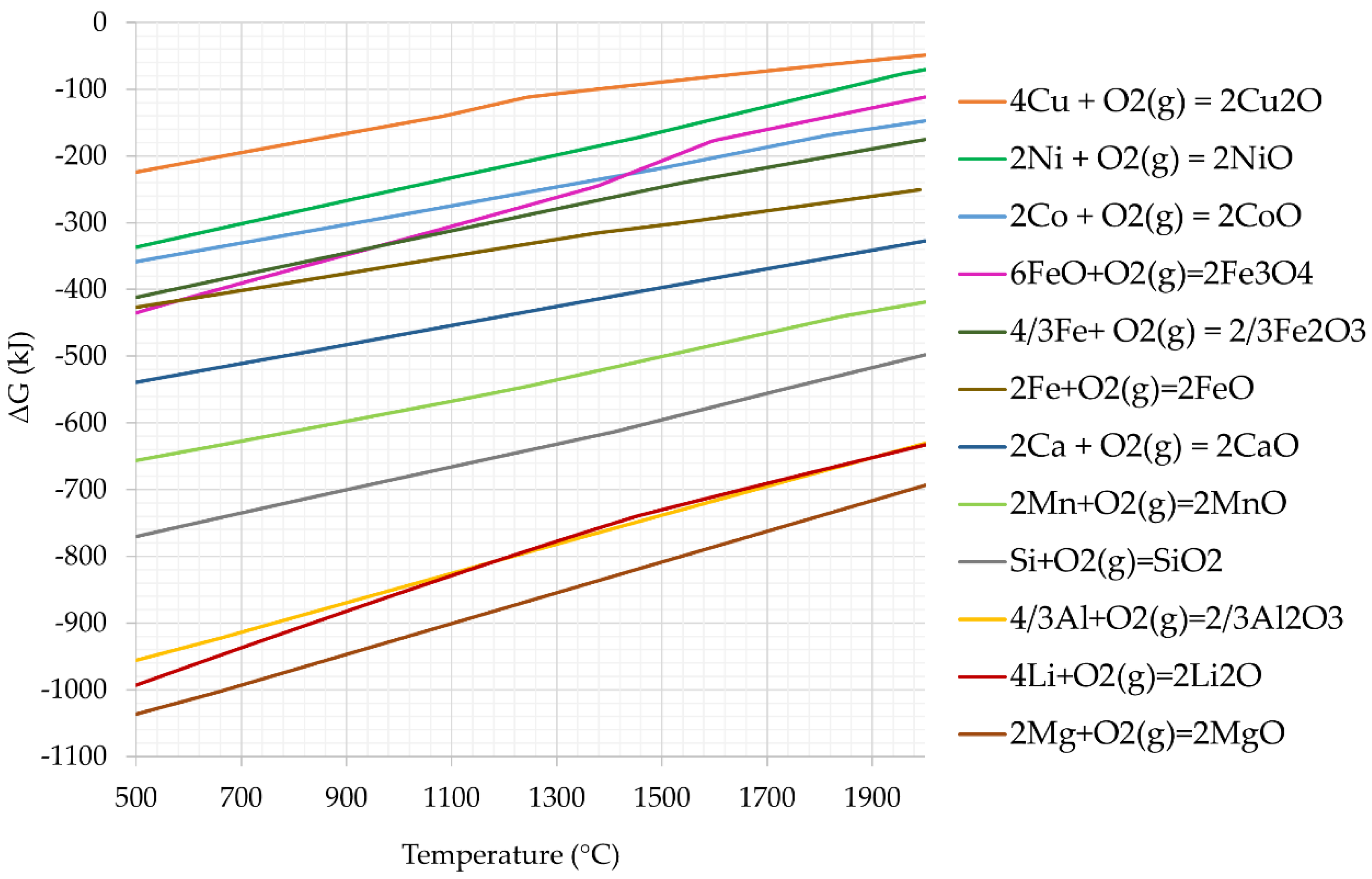
3.2.2. Chemical Reactions
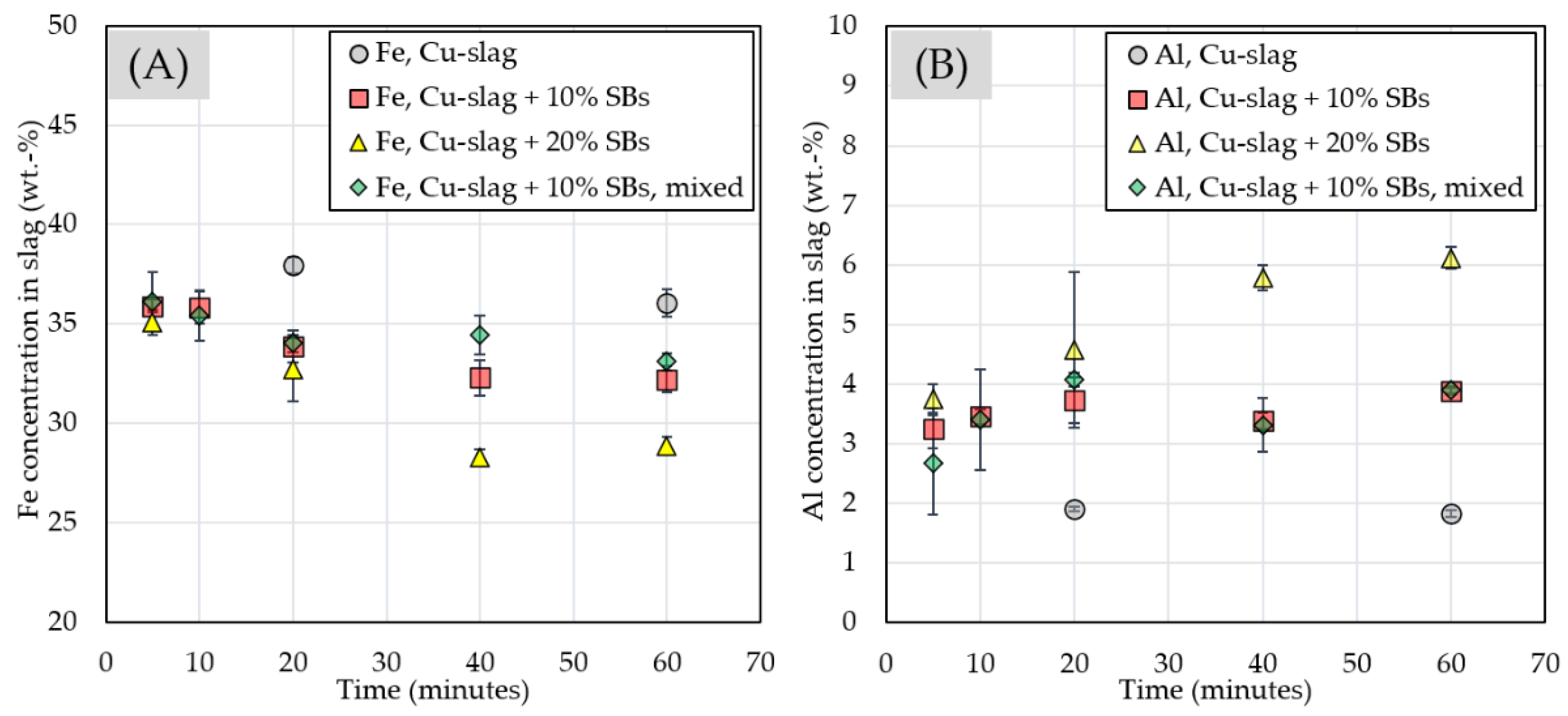
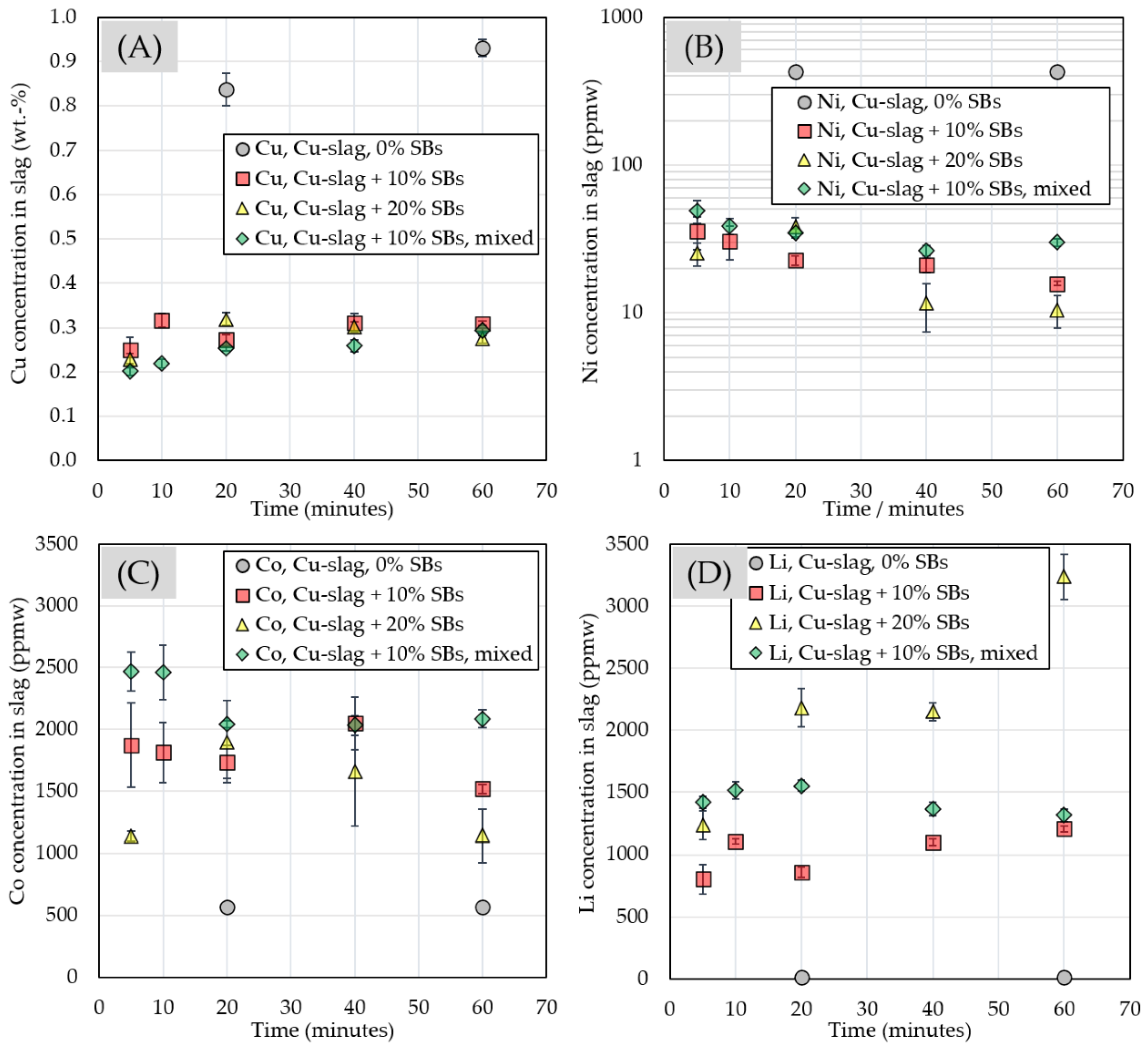
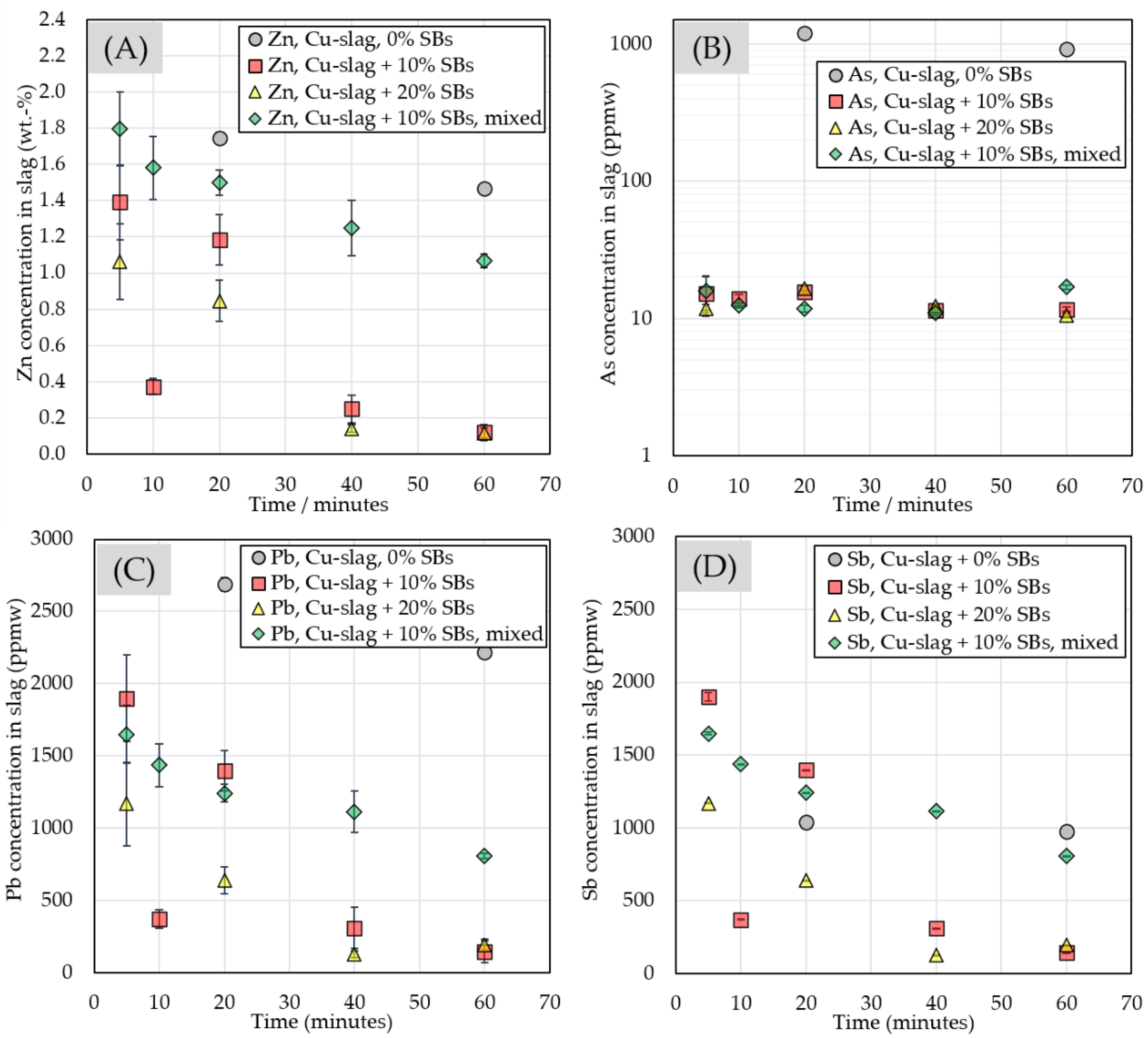
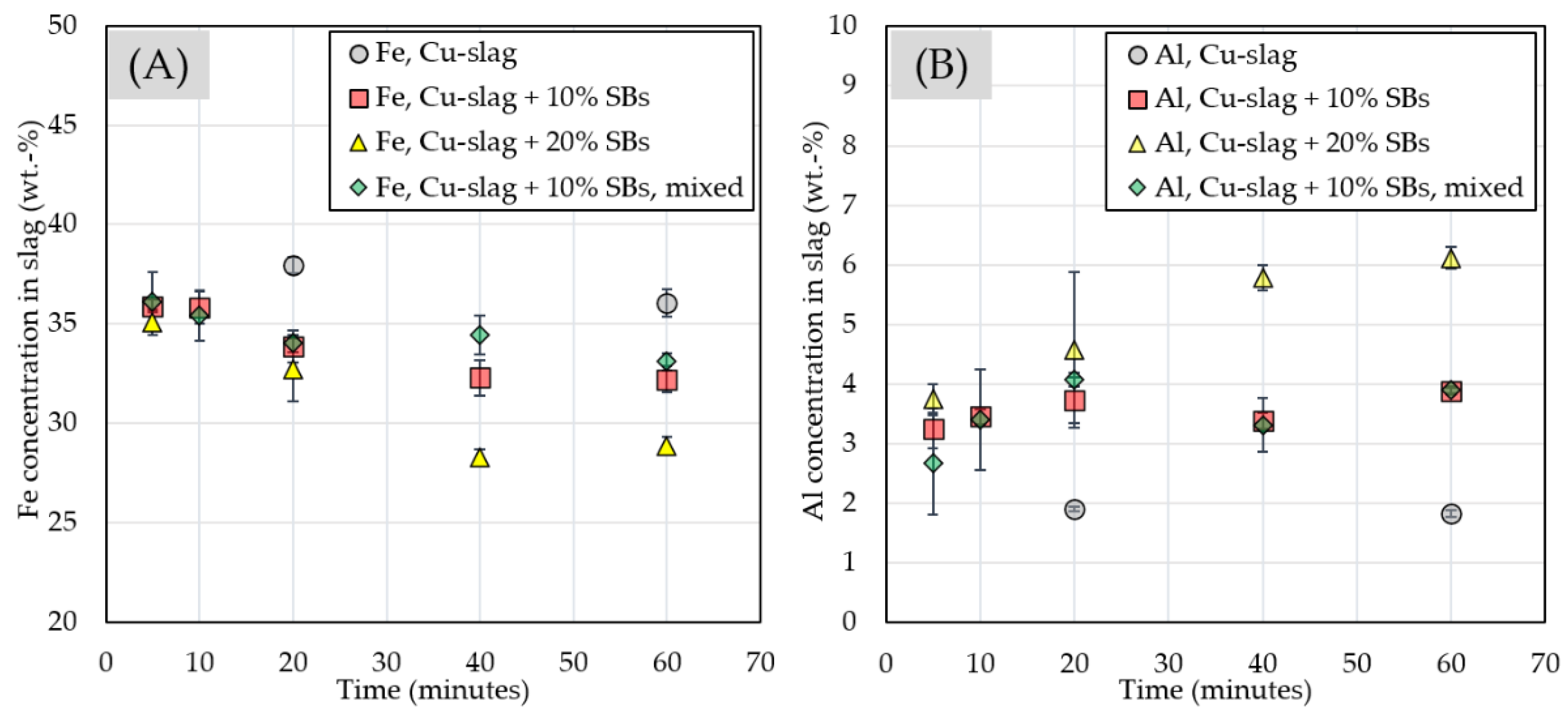
3.2.3. Chemical Composition of Slag
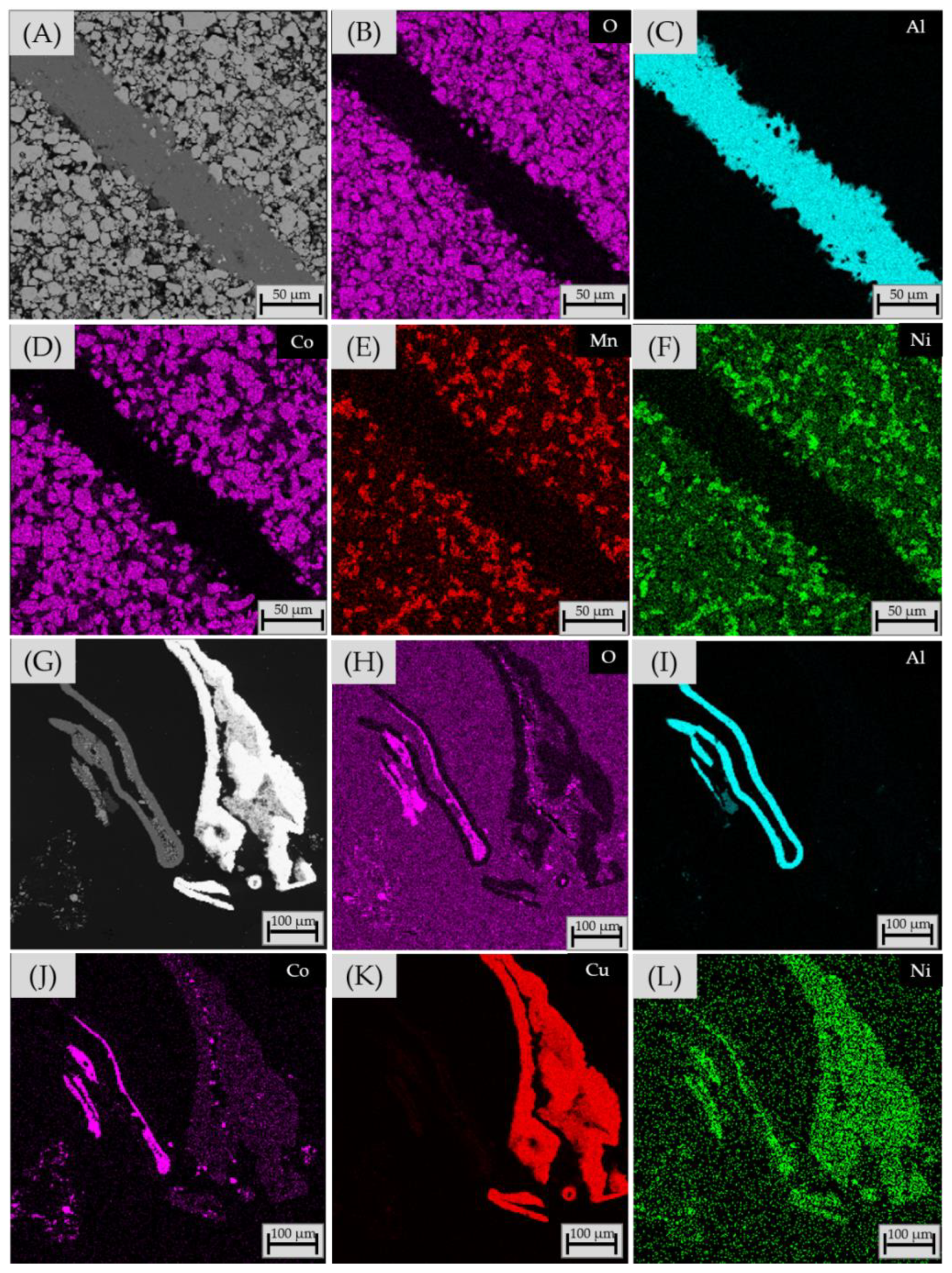
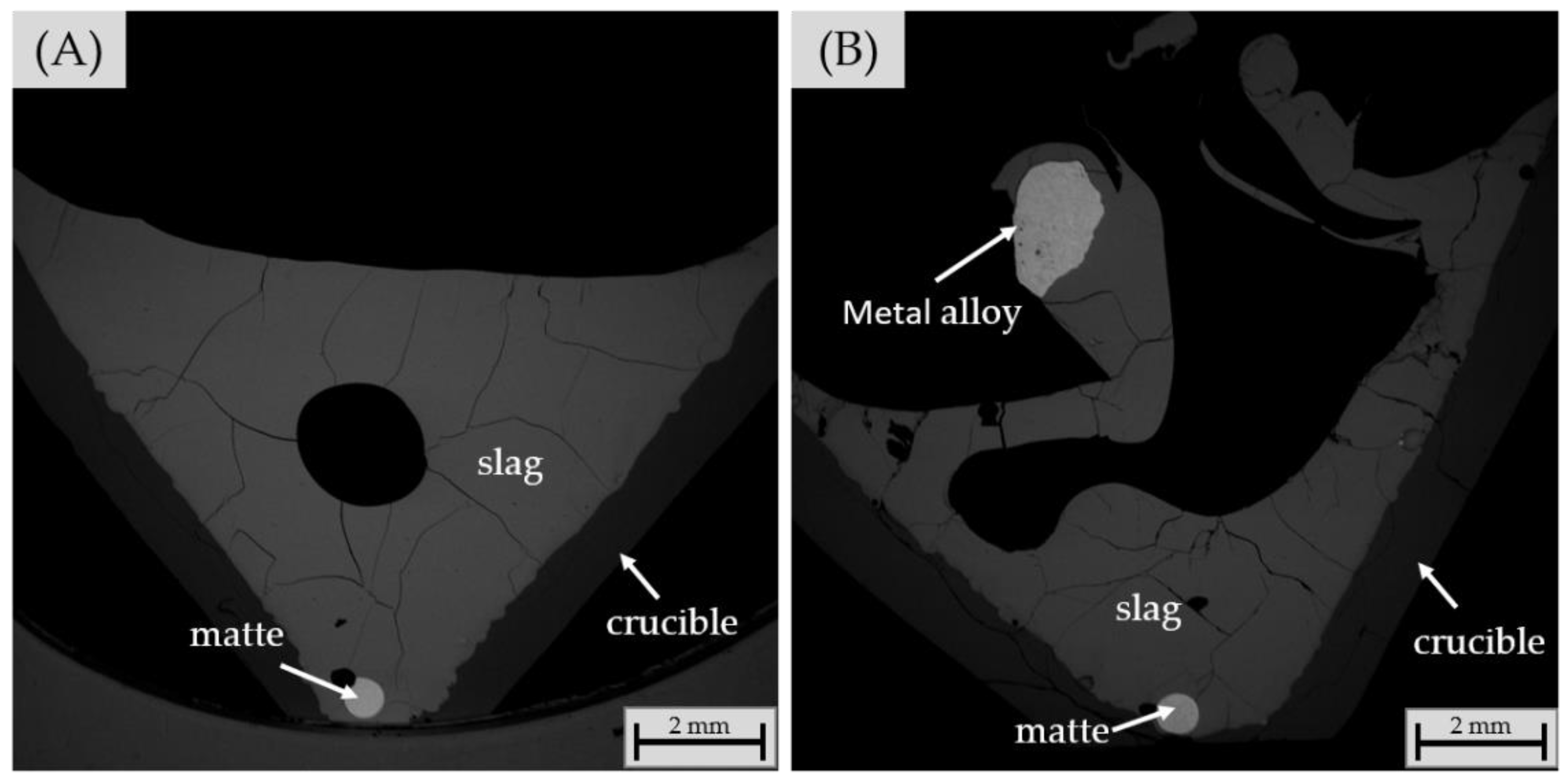
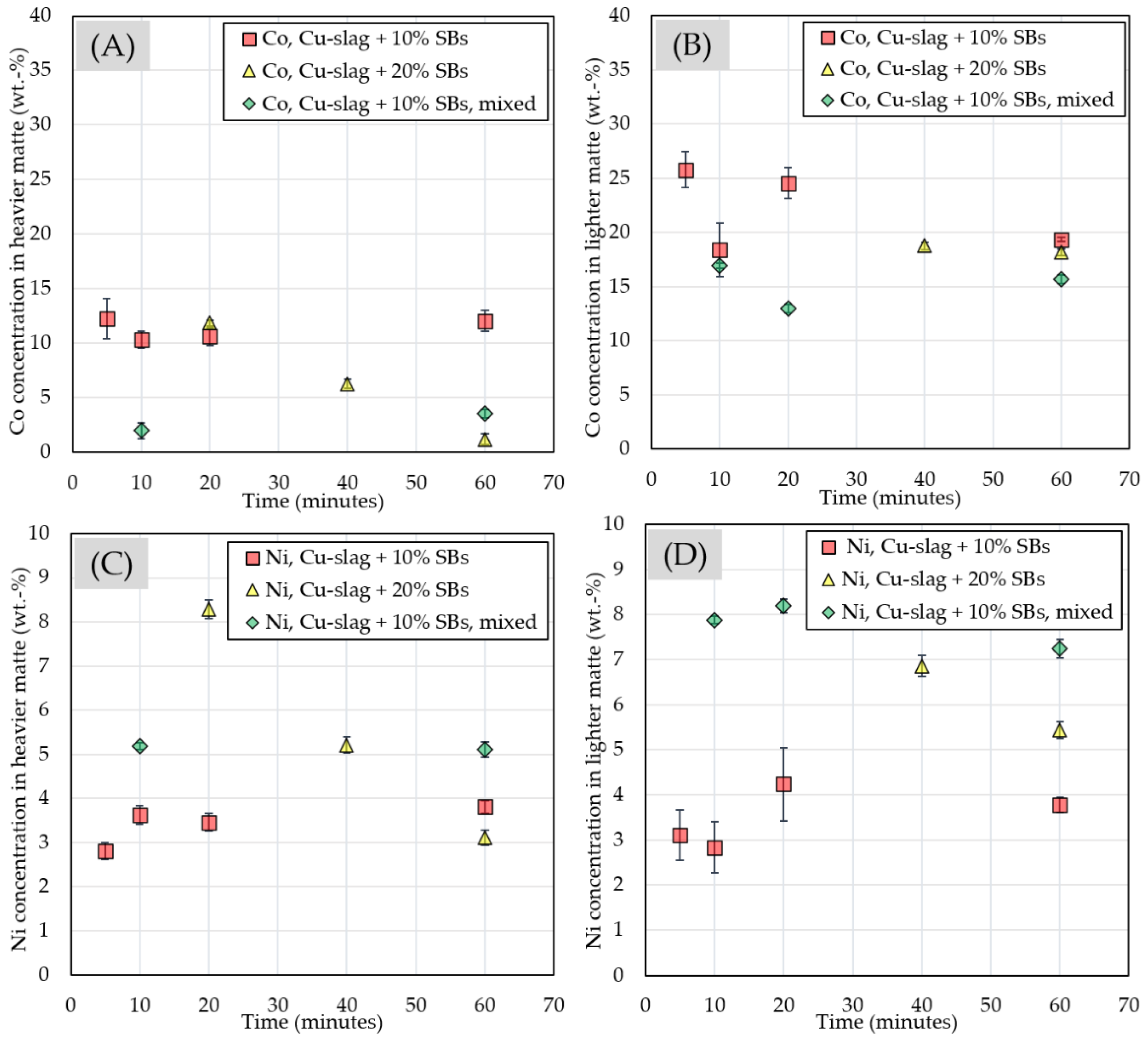
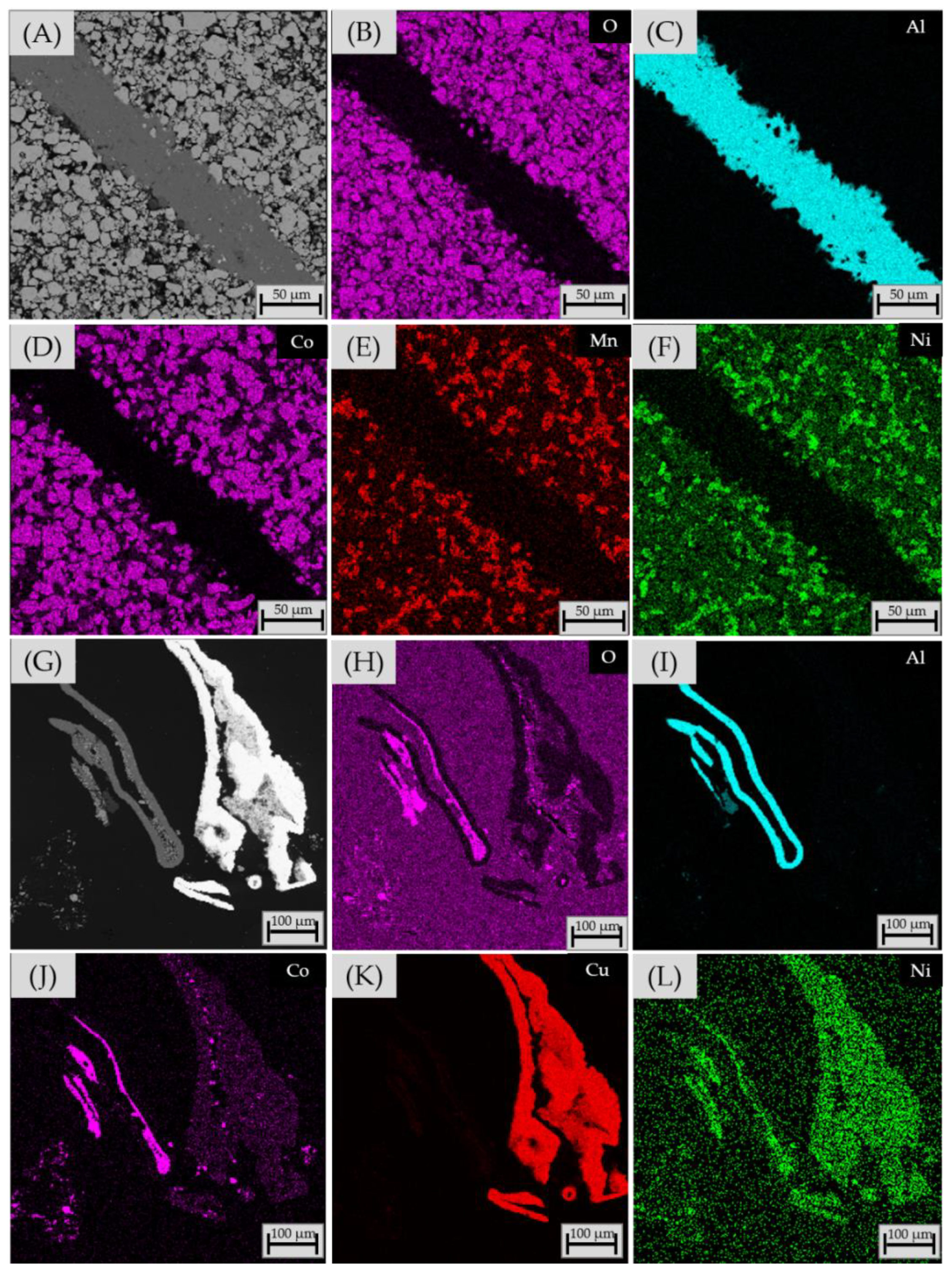
3.2.4. Chemical Composition of Matte and Alloy Phases
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Slag phase | ||||||||||
| 0% SBs | ppmw | |||||||||
| time/minutes | Cu | Zn | Co | Ni | Li | Mn | As | Sn | Sb | Pb |
| 20 | 8362 ± 361 | 17,282 ± 414 | 568 ± 21.0 | 428 ± 12.1 | 12.6 ± 0.32 | 453 ± 10.4 | 1206 ± 44.6 | 975 ± 18.9 | 1037 ± 28.4 | 2688 ± 46.7 |
| 60 | 9309 ± 194 | 14,700 ± 345 | 570 ± 21.1 | 426 ± 6.70 | 12.7 ± 0.17 | 442 ± 8.24 | 916 ± 27.7 | 910 ± 19.3 | 972 ± 16.2 | 2215 ± 35.0 |
| 10% SBs | ppmw | |||||||||
| time/minutes | Cu | Zn | Co | Ni | Li | Mn | As | Sn | Sb | Pb |
| 5 | 2487 ± 299 | 13,890 ± 2208 | 1875 ± 338 | 35.5 ± 8.92 | 804 ± 121 | 786 ± 111 | 15.2 ± 4.83 | 608 ± 84.5 | 37.2 ± 27.9 | 1899 ± 302 |
| 10 | 3172 ± 153 | 3806 ± 372 | 1814 ± 242 | 30.6 ± 7.85 | 1105 ± 18.4 | 1876 ± 44.1 | 14 ± 1.03 | 226 ± 16.2 | 11.5 ± 4.33 | 371 ± 61.9 |
| 20 | 2719 ± 119 | 11,612 ± 1405 | 1736 ± 135 | 22.6 ± 1.67 | 858 ± 41.6 | 1052 ± 40.3 | 15.6 ± 0.48 | 457 ± 30.0 | 7.30 ± 0.73 | 1396 ± 141 |
| 40 | 3104 ± 209 | 2620 ± 782 | 2049 ± 212 | 21.0 ± 2.36 | 1101 ± 27.7 | 1036 ± 84.1 | 11.5 ± 0.35 | 202 ± 68.4 | 3.31 ± 1.45 | 308 ± 142 |
| 60 | 3097 ± 42.8 | 1242 ± 462 | 1520 ± 36.2 | 15.8 ± 0.56 | 1206 ± 24.2 | 1463 ± 50.0 | 11.7 ± 0.39 | 159 ± 70.9 | 1.08 ± 0.12 | 144 ± 73.8 |
| 20% SBs | ppmw | |||||||||
| time/minutes | Cu | Zn | Co | Ni | Li | Mn | As | Sn | Sb | Pb |
| 5 | 2288 ± 124 | 10,639 ± 2159 | 1139 ± 39.2 | 25.2 ± 4.54 | 1234 ± 118 | 1257 ± 163 | 11.8 ± 0.78 | 494 ± 70.8 | 10.5 ± 3.24 | 1167 ± 286 |
| 20 | 3192 ± 142 | 8466 ± 890 | 1902 ± 332 | 38.1 ± 5.86 | 2180 ± 154 | 1954 ± 213 | 16.5 ± 0.65 | 109 ± 27.9 | 1.63 ± 0.91 | 638 ± 91.9 |
| 40 | 3019 ± 104 | 1498 ± 243 | 1661 ± 441 | 11.6 ± 4.20 | 2151 ± 71.4 | 1764 ± 134 | 12.3 ± 0.39 | 73.3 ± 18.1 | 1.16 ± 0.56 | 126 ± 23.7 |
| 60 | 2737 ± 84.4 | 1172 ± 286 | 1141 ± 214 | 10.4 ± 2.59 | 3236 ± 182 | 3053 ± 147 | 10.5 ± 0.41 | 165 ±34.4 | 0.98 ± 0.21 | 193 ± 39.1 |
| 10% SBs, mix | ppmw | |||||||||
| time/minutes | Cu | Zn | Co | Ni | Li | Mn | As | Sn | Sb | Pb |
| 5 | 2012 ± 82.1 | 17,939 ± 1973 | 2470 ± 159 | 48.7 ± 8.34 | 1418 ± 48.4 | 1173 ± 64.2 | 15.9 ± 4.51 | 494 ± 47.4 | 21.9 ± 10.3 | 1647 ± 200 |
| 10 | 2189 ± 89.7 | 15,956 ± 1897 | 2463 ± 221 | 38.7 ± 4.94 | 1516 ± 66.6 | 1260 ± 71.0 | 12.4 ± 0.49 | 446 ± 32.5 | 14.4 ± 2.83 | 1434 ± 147 |
| 20 | 2533 ± 65.7 | 14,972 ± 658 | 2045 ± 25.7 | 34.9 ± 2.38 | 1552 ± 42.5 | 1339 ± 48.2 | 11.8 ± 0.61 | 406 ± 18.8 | 6.22 ± 1.16 | 1240 ± 60.9 |
| 40 | 2592 ± 127 | 12,475 ± 1844 | 2035 ± 79.9 | 26.4 ± 2.09 | 1367 ± 55.7 | 1221 ± 52.8 | 10.9 ± 0.45 | 350 ± 30.7 | 6.21 ± 0.84 | 1113 ± 142 |
| 60 | 2936 ± 65.9 | 10,724 ± 460 | 2087 ± 74.0 | 30.0 ± 1.79 | 1317 ± 51.6 | 1437 ± 28.4 | 16.9 ± 0.65 | 344 ± 27.1 | 5.44 ± 0.34 | 809 ± 16.4 |
| Slag phase | ||||||
| 0% SBs | wt.% | |||||
| time/minutes | Al | Ca | Fe | Mg | O | Si |
| 20 | 1.90 ± 0.04 | 0.85 ± 0.04 | 37.97 ± 0.41 | 1.07 ± 0.08 | 35.67 ± 0.64 | 17.32 ± 0.23 |
| 60 | 1.83 ± 0.06 | 0.78 ± 0.04 | 36.08 ± 0.69 | 1.02 ± 0.04 | 36.90 ± 0.72 | 18.93 ± 0.63 |
| 10% SBs | wt.% | |||||
| time/minutes | Al | Ca | Fe | Mg | O | Si |
| 5 | 3.25 ± 0.33 | 0.80 ± 0.06 | 35.89 ± 0.31 | 1.06 ± 0.04 | 37.28 ± 0.22 | 16.96 ± 0.23 |
| 10 | 3.45 ± 0.12 | 0.84 ± 0.04 | 35.81 ± 0.82 | 1.15 ± 0.03 | 36.13 ± 0.66 | 18.39 ± 0.59 |
| 20 | 3.73 ± 0.40 | 0.76 ± 0.03 | 33.87 ± 0.81 | 1.08 ± 0.02 | 37.68 ± 0.32 | 18.68 ± 0.53 |
| 40 | 3.38 ± 0.13 | 0.74 ± 0.03 | 32.28 ± 0.90 | 1.04 ± 0.02 | 38.40 ± 0.46 | 20.36 ± 0.57 |
| 60 | 3.89 ± 0.06 | 0.79 ± 0.04 | 32.21 ± 0.66 | 1.05 ± 0.02 | 38.24 ± 0.25 | 20.06 ± 0.43 |
| 20% SBs | wt.% | |||||
| time/minutes | Al | Ca | Fe | Mg | O | Si |
| 5 | 3.74 ± 0.26 | 0.89 ± 0.02 | 35.07 ± 0.62 | 1.15 ± 0.03 | 37.10 ± 0.45 | 17.40 ± 0.16 |
| 20 | 4.57 ± 1.30 | 0.82 ± 0.07 | 32.69 ± 1.58 | 1.04 ± 0.14 | 36.14 ± 0.57 | 20.89 ± 0.81 |
| 40 | 5.78 ± 0.21 | 0.73 ± 0.05 | 28.28 ± 0.42 | 1.13 ± 0.03 | 38.87 ± 0.38 | 22.36 ± 0.37 |
| 60 | 6.12 ± 0.18 | 0.76 ± 0.03 | 28.87 ± 0.43 | 1.10 ± 0.04 | 37.86 ± 0.60 | 22.30 ± 0.41 |
| 10% SBs, mix | wt.% | |||||
| time/minutes | Al | Ca | Fe | Mg | O | Si |
| 5 | 2.67 ± 0.85 | 0.80 ± 0.06 | 36.13 ± 1.48 | 1.17 ± 0.03 | 36.87 ± 0.81 | 17.26 ± 0.46 |
| 10 | 3.40 ± 0.85 | 0.85 ± 0.06 | 35.43 ± 1.26 | 1.12 ± 0.13 | 36.64 ± 0.81 | 18.13 ± 0.66 |
| 20 | 4.08 ± 0.12 | 0.93 ± 0.02 | 34.02 ± 0.44 | 1.13 ± 0.02 | 36.55 ± 0.39 | 19.28 ± 0.21 |
| 40 | 3.31 ± 0.45 | 0.83 ± 0.09 | 34.45 ± 0.96 | 1.06 ± 0.05 | 36.75 ± 0.56 | 19.75 ± 0.42 |
| 60 | 3.90 ± 0.06 | 0.82± 0.06 | 33.11 ± 0.43 | 1.09 ± 0.02 | 36.72 ± 0.76 | 20.67 ± 0.31 |
| Heavier matte phase | |||||||||
| 0% SBs | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 20 | ˂dl | 70.2 ± 0.31 | 3.91 ± 0.02 | 0.24 ± 0.01 | ˂dl | 0.06 ± 0.04 | ˂dl | 0.51 ± 0.05 | 20.1 ± 0.01 |
| 60 | ˂dl | 78.4 ± 5.88 | 2.94 ± 1.06 | 0.30 ± 0.25 | ˂dl | 0.06 ± 0.05 | ˂dl | 1.00 ± 0.64 | 14.4 ± 8.02 |
| 10% SBs | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | 12.2 ± 1.85 | 49.3 ± 4.97 | 24.7 ± 3.46 | 2.81 ± 0.19 | 3.10 ± 0.47 | 1.27 ± 0.29 | 2.11 ± 0.47 | 0.71 ± 0.17 | 2.68 ± 1.19 |
| 10 | 10.3 ± 0.77 | 52.5 ± 2.18 | 23.2 ± 1.57 | 3.63 ± 0.21 | 2.88 ± 0.23 | 1.40 ± 0.17 | 1.62 ± 0.24 | 0.67 ± 0.17 | 2.43 ± 0.88 |
| 20 | 10.6 ± 0.81 | 50.3 ± 2.21 | 25.9 ± 1.77 | 3.46 ± 0.20 | 2.44 ± 0.40 | 1.26 ± 0.28 | 1.46 ± 0.34 | 0.36 ± 0.11 | 2.90 ± 1.43 |
| 40 | - | - | - | - | - | - | - | - | - |
| 60 | 12.1 ± 0.95 | 31.4 ± 2.39 | 38.2 ± 2.68 | 3.81 ± 0.14 | 4.43 ± 0.71 | 2.18 ± 0.23 | 2.39 ± 0.34 | 1.65 ± 0.27 | 2.83 ± 0.44 |
| 20% SBs | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | - | - | - | - | - | - | - | - | - |
| 20 | 11.8 ± 0.28 | 40.9 ± 1.22 | 29.6 ± 0.73 | 8.28 ± 0.21 | 2.26 ± 0.18 | 1.25 ± 0.08 | 0.72 ± 0.05 | 1.00 ± 0.14 | 2.74 ± 0.26 |
| 40 | 6.28 ± 0.42 | 58.8 ± 1.83 | 23.0 ± 1.42 | 5.21 ± 0.18 | 1.64 ± 0.11 | 1.02 ± 0.07 | 1.36 ± 0.05 | 0.20 ± 0.06 | 2.25 ± 0.33 |
| 60 | 1.21 ± 0.47 | 69.4 ± 1.38 | 3.84 ± 1.33 | 3.11 ± 0.18 | 3.95 ± 0.84 | 12.0 ± 1.54 | 4.52 ± 0.44 | 0.30 ± 0.09 | 0.43 ± 0.29 |
| 10% SBs, mix | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | - | - | - | - | - | - | - | - | - |
| 10 | 2.00 ± 0.71 | 59.2 ± 2.01 | 4.00 ± 1.68 | 5.19 ± 0.08 | 6.55 ± 0.52 | 17.5 ± 1.61 | 3.33 ± 0.22 | 0.99 ± 0.15 | 0.69 ± 0.32 |
| 20 | - | - | - | - | - | - | - | - | - |
| 40 | - | - | - | - | - | - | - | - | - |
| 60 | 3.56 ± 0.38 | 55.5 ± 1.63 | 9.74 ± 1.08 | 5.11 ± 0.17 | 5.21 ± 0.20 | 14.5 ± 0.51 | 3.45 ± 0.31 | 0.84 ± 0.13 | 0.99 ± 0.20 |
| Lighter matte phase | |||||||||
| 10% SBs | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | 25.8 ± 1.65 | 11.7 ± 0.24 | 55.8 ± 1.67 | 3.11 ± 0.56 | 0.43 ± 0.11 | 0.08 ± 0.05 | 0.13 ± 0.08 | 1.71 ± 0.18 | 0.76 ± 0.11 |
| 10 | 18.4 ± 2.46 | 8.80 ± 2.17 | 66.4 ± 4.20 | 2.83 ± 0.57 | 0.57 ± 0.07 | 0.06 ± 0.04 | ˂dl | 1.48 ± 0.21 | 0.70 ± 0.12 |
| 20 | 24.6 ± 1.41 | 11.6 ± 0.19 | 56.4 ± 0.83 | 4.24 ± 0.82 | 0.42 ± 0.04 | 0.06 ± 0.05 | ˂dl | 1.38 ± 0.14 | 0.57 ± 0.05 |
| 40 | - | - | - | - | - | - | - | - | - |
| 60 | 19.4 ± 0.22 | 8.97 ± 0.26 | 62.3 ± 0.46 | 3.78 ± 0.17 | 0.85 ± 0.04 | 0.14 ± 0.03 | 0.18 ± 0.05 | 2.68 ± 0.16 | 1.12 ± 0.03 |
| 20% SBs | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | - | - | - | - | - | - | - | - | - |
| 20 | - | - | - | - | - | - | - | - | - |
| 40 | 18.8 ± 0.36 | 11.7 ± 0.38 | 61.3 ± 0.46 | 6.86 ± 0.23 | 0.32 ± 0.05 | ˂dl | 0.05 ± 0.04 | 0.20 ± 0.13 | 0.36 ± 0.03 |
| 60 | 18.1 ± 0.30 | 8.79 ± 0.74 | 51.3 ± 0.55 | 5.44 ± 0.18 | 8.71 ± 0.32 | 1.43 ± 0.15 | 0.48 ± 0.32 | 2.19 ± 0.21 | 2.41 ± 0.14 |
| 10% SBs, mix | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | - | - | - | - | - | - | - | - | - |
| 10 | 17.0 ± 0.24 | 11.2 ± 0.77 | 38.0 ± 0.65 | 7.88 ± 0.08 | 16.1 ± 0.20 | 4.03 ± 0.26 | 0.78 ± 0.09 | 2.21 ± 0.12 | 1.80 ± 0.12 |
| 20 | 13.0 ± 0.36 | 17.4 ± 0.93 | 36.2 ± 0.82 | 8.19 ± 0.16 | 11.1 ± 0.32 | 6.44 ± 0.47 | 1.31 ± 0.09 | 2.84 ± 0.09 | 2.02 ± 0.19 |
| 40 | - | - | - | - | - | - | - | - | - |
| 60 | 15.7 ± 0.35 | 10.1 ± 0.91 | 43.3 ± 0.65 | 7.24 ± 0.20 | 11.4 ± 0.21 | 3.31 ± 0.27 | 0.76 ± 0.08 | 4.04 ± 0.11 | 2.88 ± 0.13 |
| Metal alloy phase | |||||||||
| 10% SBs | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | - | - | - | - | - | - | - | - | - |
| 10 | 10.3 | 22.2 | 59.1 | 2.71 | 1.69 | 1.06 | 0.79 | 0.82 | 0.62 |
| 20 | 11.9 | 26.8 | 54.4 | 2.55 | 1.18 | 0.84 | 0.82 | 0.26 | 0.41 |
| 40 | 14.5 | 26.3 | 51.5 | 2.15 | 1.75 | 0.82 | 0.72 | 0.94 | 0.52 |
| 60 | 13.9 | 29.8 | 46.6 | 3.33 | 2.25 | 1.38 | 0.86 | 0.78 | 0.41 |
| 20% SBs | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | 18.6 | 30.1 | 43.7 | 3.86 | 0.86 | 0.58 | 0.91 | 0.28 | 0.26 |
| 20 | 18.9 | 26.5 | 50.7 | 1.59 | 0.74 | 0.35 | 0.34 | 0.21 | 0.38 |
| 40 | 15.2 | 24.7 | 54.6 | 3.20 | 0.35 | 0.38 | 0.36 | 0.44 | 0.39 |
| 60 | 13 | 23.2 | 57.9 | 2.57 | 1.05 | 0.66 | 0.54 | 0.12 | 0.30 |
| 10% SBs, mix | wt.% | ||||||||
| time/minutes | Co | Cu | Fe | Ni | As | Sb | Sn | Pb | S |
| 5 | 14.8 | 28.9 | 49.0 | 2.58 | 1.40 | 0.76 | 0.88 | 0.66 | 0.30 |
| 10 | 14.8 | 27.0 | 48.8 | 2.52 | 2.15 | 1.11 | 1.04 | 0.91 | 0.52 |
| 20 | 14.5 | 26.9 | 50.4 | 3.25 | 1.40 | 0.81 | 0.91 | 0.88 | 0.42 |
| 40 | 16.0 | 20.4 | 55.0 | 2.79 | 1.33 | 0.68 | 0.54 | 0.84 | 0.40 |
| 60 | 13.0 | 29.9 | 47.1 | 3.29 | 2.02 | 1.14 | 0.96 | 0.82 | 0.37 |
References
- Velázquez-Martínez, O.; Valio, J.; Santasalo-Aarnio, A.; Reuter, M.; Serna-Guerrero, R. A Critical Review of Lithium-Ion Battery Recycling Processes from a Circular Economy Perspective. Batteries 2019, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Wang, Z.; Cao, H.; Sun, Y.; Zhang, Y.; Sun, Z.H. A Critical Review and Analysis on the Recycling of Spent Lithium-Ion Batteries. ACS Sustain. Chem. Eng. 2018, 6, 1504–1521. [Google Scholar] [CrossRef]
- Pinegar, H.; Smith, Y.R. Mechanical Beneficiation of End-of-Life Lithium-Ion Battery Components. In Proceedings of the Energy Technology 2020: Recycling, Carbon Dioxide Management, and Other Technologies, San Diego, CA, USA, 23–27 February 2020; Chen, X., Zhong, Y., Zhang, L., Howarter, J.A., Baba, A.A., Wang, C., Sun, Z., Zhang, M., Olivetti, E., Luo, A., et al., Eds.; Springer: Cham, Switzerland, 2020; pp. 259–267. [Google Scholar]
- Zhang, G.; Yuan, X.; He, Y.; Wang, H.; Zhang, T.; Xie, W. Recent advances in pretreating technology for recycling valuable metals from spent lithium-ion batteries. J. Hazard. Mater. 2021, 406, 124332. [Google Scholar] [CrossRef]
- Velázquez-Martinez, O.; Porvali, A.; van den Boogaart, K.G.; Santasalo-Aarnio, A.; Lundström, M.; Reuter, M.; Serna-Guerrero, R. On the Use of Statistical Entropy Analysis as Assessment Parameter for the Comparison of Lithium-Ion Battery Recycling Processes. Batteries 2019, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Harper, G.; Sommerville, R.; Kendrick, E.; Driscoll, L.; Slater, P.; Stolkin, R.; Walton, A.; Christensen, P.; Heidrich, O.; Lambert, S.; et al. Recycling lithium-ion batteries from electric vehicles. Nature 2019, 575, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Brückner, L.; Frank, J.; Elwert, T. Industrial Recycling of Lithium-Ion Batteries—A Critical Review of Metallurgical Process Routes. Metals 2020, 10, 1107. [Google Scholar] [CrossRef]
- Widijatmoko, S.D.; Fu, G.; Wang, Z.; Hall, P. Recovering lithium cobalt oxide, aluminium, and copper from spent lithium-ion battery via attrition scrubbing. J. Clean. Prod. 2020, 260, 120869. [Google Scholar] [CrossRef]
- Wang, X.; Gaustad, G.; Babbitt, C.W. Targeting high value metals in lithium-ion battery recycling via shredding and size-based separation. Waste Manag. 2016, 51, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommerville, R.; Shaw-Stewart, J.; Goodship, V.; Rowson, N.; Kendrick, E. A review of physical processes used in the safe recycling of lithium ion batteries. Sustain. Mater. Technol. 2020, 25, e00197. [Google Scholar] [CrossRef]
- Zhang, G.; Du, Z.; He, Y.; Wang, H.; Xie, W.; Zhang, T. A Sustainable Process for the Recovery of Anode and Cathode Materials Derived from Spent Lithium-Ion Batteries. Sustainability 2019, 11, 2363. [Google Scholar] [CrossRef] [Green Version]
- Pinegar, H.; Smith, Y.R. Recycling of End-of-Life Lithium Ion Batteries, Part I: Commercial Processes. J. Sustain. Metall. 2019, 5, 402–416. [Google Scholar] [CrossRef]
- Widijatmoko, S.D.; Gu, F.; Wang, Z.; Hall, P. Selective liberation in dry milled spent lithium-ion batteries. Sustain. Mater. Technol. 2019, 23, e00134. [Google Scholar] [CrossRef]
- Yu, J.; Tan, Q.; Li, J. Exploring a green route for recycling spent lithium-ion batteries: Revealing and solving deep screening problem. J. Clean. Prod. 2020, 255, 120269. [Google Scholar] [CrossRef]
- Yu, J.; He, Y.; Ge, Z.; Li, H.; Xie, W.; Wang, S. A promising physical method for recovery of LiCoO2 and graphite from spent lithium-ion batteries: Grinding flotation. Sep. Purif. Technol. 2018, 190, 45–52. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, T.; He, Y.; Zhao, Y.; Wang, S.; Zhang, G.; Zhang, Y.; Feng, Y. Recovery of valuable materials from spent lithium-ion batteries by mechanical separation and thermal treatment. J. Clean. Prod. 2018, 185, 646–652. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Hu, T.; Bai, X.; Wang, S.; Xie, W.; Hao, J.; He, Y. Recovery of LiCoO2 and graphite from spent lithium-ion batteries by cryogenic grinding and froth flotation. Miner. Eng. 2020, 148, 106223. [Google Scholar] [CrossRef]
- Zhang, G.; He, Y.; Feng, Y.; Wang, H.; Zhu, X. Pyrolysis-Ultrasonic-Assisted Flotation Technology for Recovering Graphite and LiCoO2 from Spent Lithium-Ion Batteries. ACS Sustain. Chem. Eng. 2018, 6, 10896–10904. [Google Scholar] [CrossRef]
- Zhang, G.; He, Y.; Wang, H.; Feng, Y.; Xie, W.; Zhu, X. Application of mechanical crushing combined with pyrolysis-enhanced flotation technology to recover graphite and LiCoO2 from spent lithium-ion batteries. J. Clean. Prod. 2019, 231, 1418–1427. [Google Scholar] [CrossRef]
- He, Y.; Zhang, T.; Wang, F.; Zhang, G.; Zhang, W.; Wang, J. Recovery of LiCoO2 and graphite from spent lithium-ion batteries by Fenton reagent-assisted flotation. J. Clean. Prod. 2017, 143, 319–325. [Google Scholar] [CrossRef]
- Ruismäki, R.; Rinne, T.; Dańczak, A.; Taskinen, P.; Serna-Guerrero, R.; Jokilaakso, A. Integrating Flotation and Pyrometallurgy for Recovering Graphite and Valuable Metals from Battery Scrap. Metals 2020, 10, 680. [Google Scholar] [CrossRef]
- Dańczak, A.; Ruismäki, R.; Rinne, T.; Klemettinen, L.; O’Brien, H.; Taskinen, P.; Jokilaakso, A.; Serna-Guerrero, R. Worth from Waste: Utilizing a Graphite-Rich Fraction from Spent Lithium-Ion Batteries as Alternative Reductant in Nickel Slag Cleaning. Minerals 2021, 11, 784. [Google Scholar] [CrossRef]
- Gorai, B.; Jana, R. Premchand Characteristics and utilisation of copper slag—A review. Resour. Conserv. Recycl. 2003, 39, 299–313. [Google Scholar] [CrossRef]
- Potysz, A.; van Hullebusch, E.D.; Kierczak, J.; Grybos, M.; Lens, P.N.L.; Guibaud, G. Copper Metallurgical Slags—Current Knowledge and Fate: A Review. Crit. Rev. Environ. Sci. Technol. 2015, 45, 2424–2488. [Google Scholar] [CrossRef]
- Alp, I.; Deveci, H.; Süngün, H. Utilization of flotation wastes of copper slag as raw material in cement production. J. Hazard. Mater. 2008, 159, 390–395. [Google Scholar] [CrossRef]
- Heo, J.H.; Chung, Y.; Park, J.H. Recovery of iron and removal of hazardous elements from waste copper slag via a novel aluminothermic smelting reduction (ASR) process. J. Clean. Prod. 2016, 137, 777–787. [Google Scholar] [CrossRef]
- Messler, R. Aluminothermic Welding. In Principles of Welding: Processes, Physics, Chemistry, and Metallurgy, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 1999. [Google Scholar]
- Vanderbruggen, A.; Sygusch, J.; Rudolph, M.; Serna-Guerrero, R. A contribution to understanding the flotation behavior of lithium metal oxides and spheroidized graphite for lithium-ion battery recycling. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 127111. [Google Scholar] [CrossRef]
- Nuorivaara, T.; Serna-Guerrero, R. Amphiphilic cellulose and surfactant mixtures as green frothers in mineral flotation. 1. Characterization of interfacial and foam stabilization properties. Colloids Surf. A Physicochem. Eng. Asp. 2020, 604, 125297. [Google Scholar] [CrossRef]
- Nuorivaara, T.; Serna-Guerrero, R. Unlocking the potential of sustainable chemicals in mineral processing: Improving sphal-erite flotation using amphiphilic cellulose and frother mixtures. J. Clean. Prod. 2020, 261, 121143. [Google Scholar] [CrossRef]
- Wills, B.A.; Finch, J. Wills’ Mineral Processing Technology: An Introduction to the Practical Aspects of Ore Treatment and Mineral Recovery, 8th ed.; Butterworth-Heinemann: Oxford, UK, 2015; pp. 1–27. [Google Scholar]
- Dańczak, A.; Klemettinen, L.; O’Brien, H.; Taskinen, P.; Lindberg, D.; Jokilaakso, A. Slag Chemistry and Behavior of Nickel and Tin in Black Copper Smelting with Alumina and Magnesia-Containing Slags. J. Sustain. Met. 2021, 7, 1–14. [Google Scholar] [CrossRef]
- Van Achterberg, E.; Ryan, C.G.; Jackson, S.E.; Griffin, W.L. Data reduction software for LA-ICP-MS: Appendix. In Laser Ablation-ICP-Mass Spectrometry in the Earth Sciences: Principles and Applications; Sylvester, P.J., Ed.; Short Course Series; Mineralogical Association of Canada: Ottawa, ON, Canada, 2001; pp. 239–243. [Google Scholar]
- Jochum, K.P.; Weis, U.; Stoll, B.; Kuzmin, D.; Yang, Q.; Raczek, I.; Jacob, D.E.; Stracke, A.; Birbaum, K.; Frick, D.A.; et al. Determination of Reference Values for NIST SRM 610-617 Glasses Following ISO Guidelines. Geostand. Geoanal. Res. 2011, 35, 397–429. [Google Scholar] [CrossRef]
- Vanderbruggen, A.; Gugala, E.; Blannin, R.; Bachmann, K.; Serna-Guerrero, R.; Rudolph, M. Automated mineralogy as a novel approach for the compositional and textural characterization of spent lithium-ion batteries. Miner. Eng. 2021, 169, 106924. [Google Scholar] [CrossRef]
- Vignes, A. Extractive Metallurgy 2: Metallurgical Reaction Processes, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 9781848212879. [Google Scholar]
- Avarmaa, K.; Järvenpää, M.; Klemettinen, L.; Marjakoski, M.; Taskinen, P.; Lindberg, D.; Jokilaakso, A. Battery Scrap and Biochar Utilization for Improved Metal Recoveries in Nickel Slag Cleaning Conditions. Batteries 2020, 6, 58. [Google Scholar] [CrossRef]







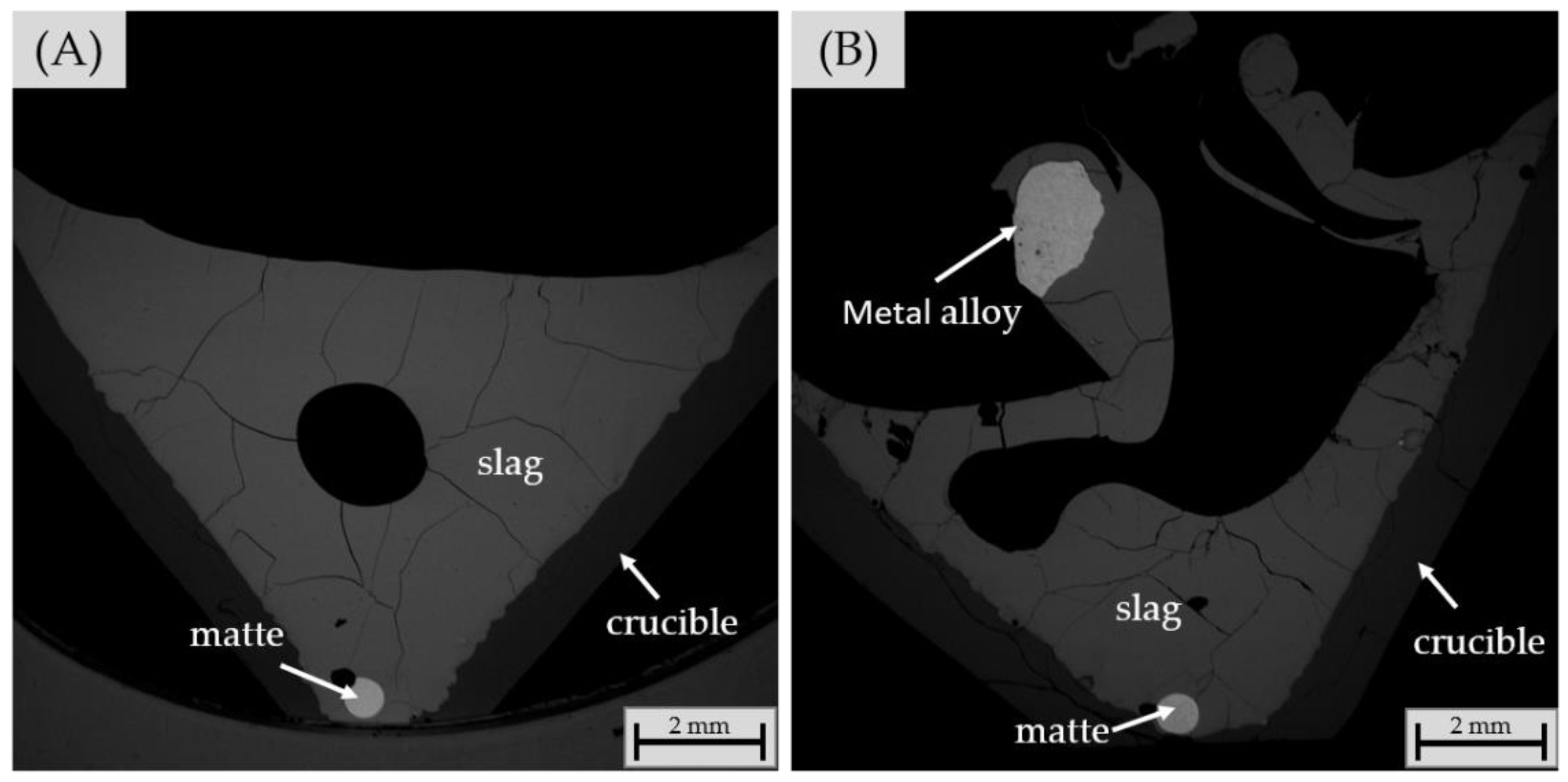

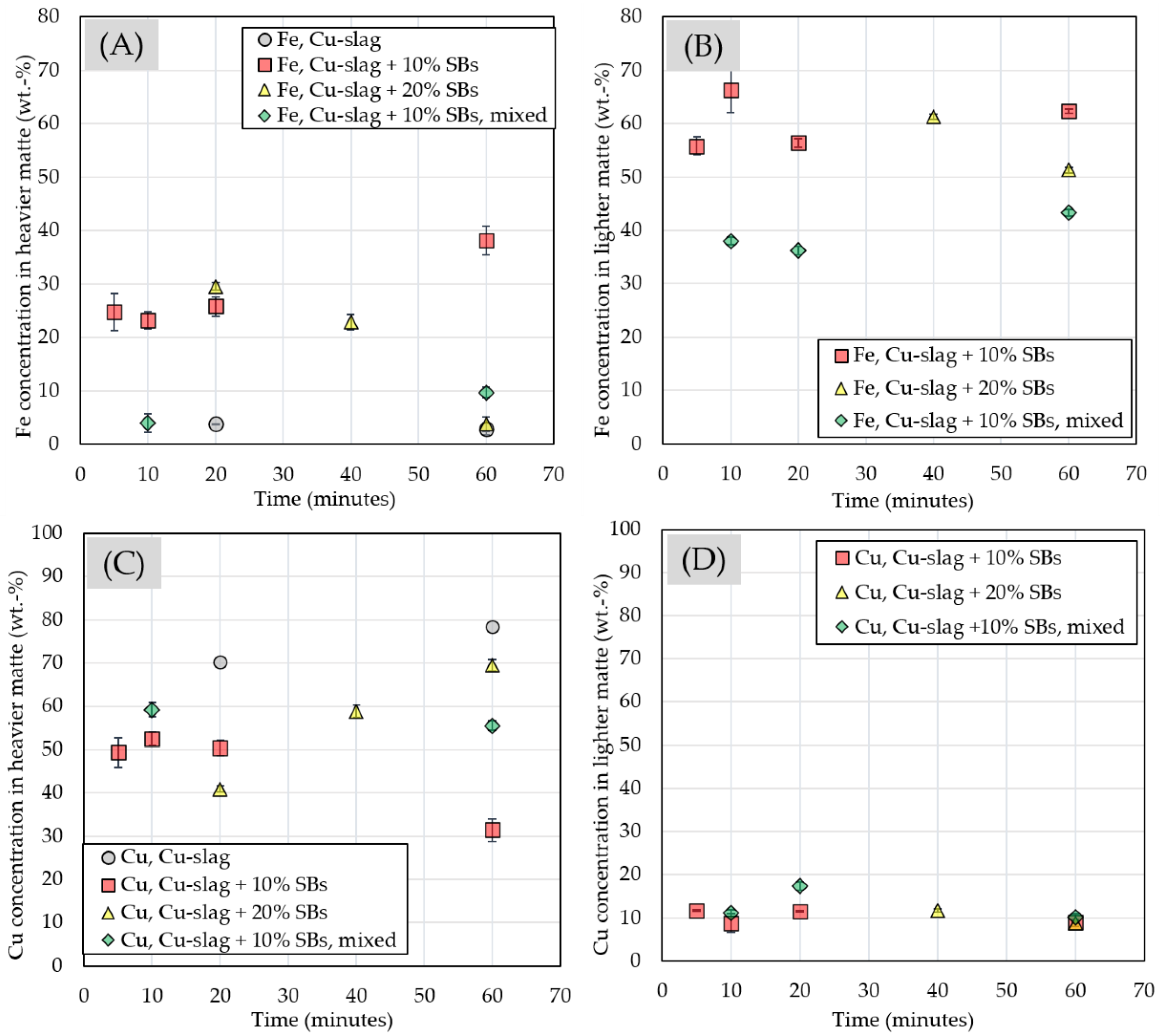
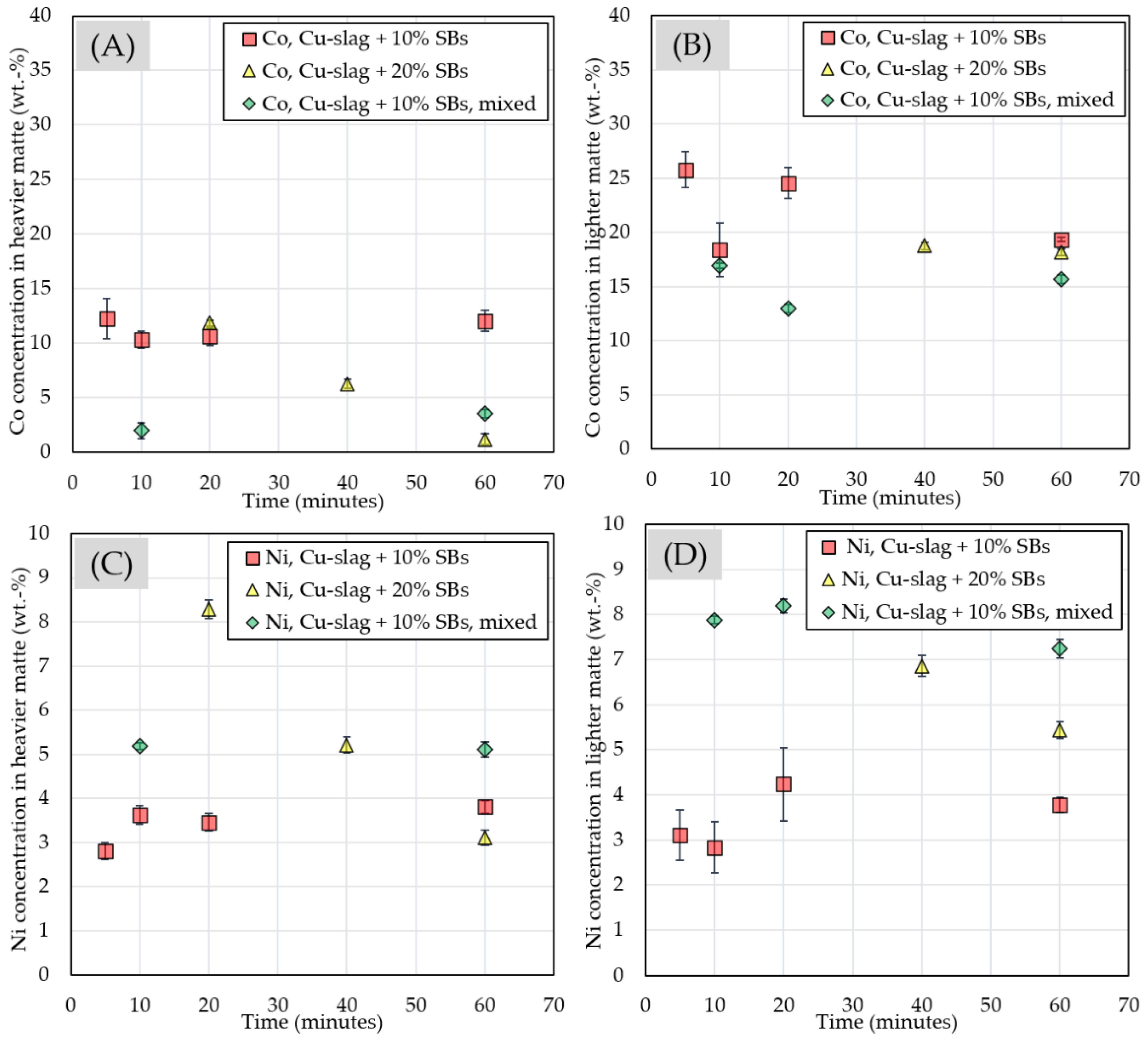
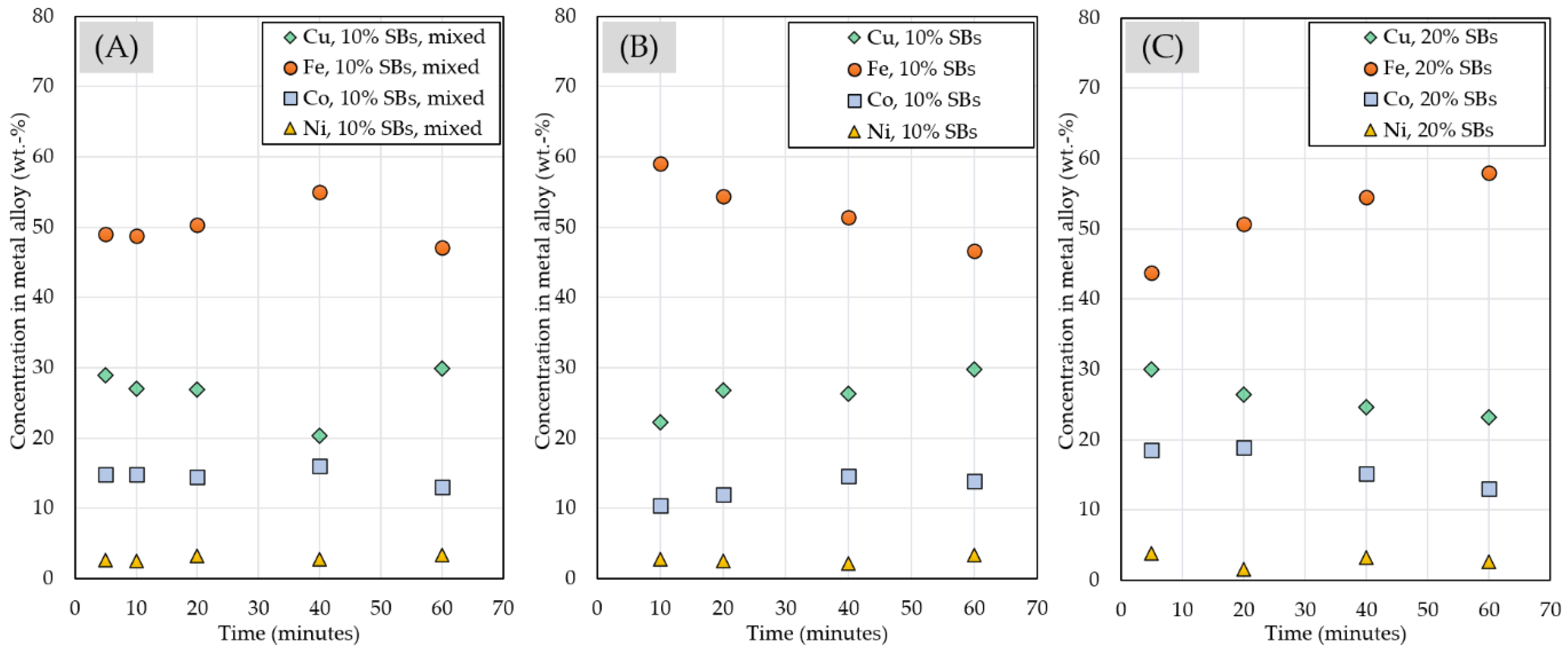
| Experiment | Frother | Collector |
|---|---|---|
| CF1 | CellFroth 30 ppm | no collector |
| CF2 | CellFroth 30 ppm | kerosene 150 g/t |
| CF3 | CellFroth 30 ppm | kerosene 300 g/t |
| CF4 | HPMC 30 ppm | No collector |
| MIBC1 | MIBC 8 ppm | kerosene 150 g/t |
| MIBC2 | MIBC 8 ppm | kerosene 150 g/t |
| MIBC3 | MIBC 8 ppm | kerosene 150 g/t |
| MIBC4 | MIBC 8 ppm | kerosene 150 g/t |
| Component | wt.% |
|---|---|
| Na | 0.416 |
| Mg | 1.19 |
| Al | 2.14 |
| K | 0.893 |
| Ca | 0.805 |
| Cr | 0.034 |
| Fe 1 | 38.3 |
| Co | 0.062 |
| Ni | 0.048 |
| Cu | 1.19 |
| Zn | 1.78 |
| As | 0.19 |
| Sb | 0.10 |
| Pb | 0.309 |
| S | 0.47 |
| SiO2 | 33 |
| Fe3O4 | 11.7 |
| Cu | Co | Li | Ni | Mn | Pb | Sn | Zn | S | As | Sb | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| External standard for slag | NIST610 [34] | ||||||||||
| Internal standard for slag | 29Si | ||||||||||
| Detection limit for slag (ppmw) for the isotope used | 65Cu: 0.018 | 59Co: 0.017 | 7Li: 0.043 | 60Ni: 0.071 | 55Mn: 0.020 | 208Pb: 0.002 | 120Sn: 0.025 | 67Zn: 0.228 | 34S: 3.440 | 75As: 0.081 | 121Sb: 0.007 |
| CF1 | CF2 | CF3 | CF4 | MIBC1 | MIBC2 | MIBC3 | MIBC4 | |
|---|---|---|---|---|---|---|---|---|
| Co (wt.%) | 18.7 | 19.1 | 19.5 | 20.3 | 19.2 | 18.0 | 21.6 | 18.7 |
| Ni (wt.%) | 2.1 | 2.2 | 2.2 | 2.3 | 2.2 | 2.1 | 2.5 | 2.2 |
| Mn (wt.%) | 2.0 | 2.0 | 2.0 | 2.1 | 2.0 | 1.9 | 2.2 | 1.9 |
| Cu (wt.%) | 26.4 | 26.1 | 23.5 | 21.7 | 27.4 | 22.0 | 18.1 | 23.4 |
| Graphite (average) (wt.%) | 19.85 | |||||||
| Element | Al | Co | Cu | Fe | Li | Mn | Ni | P | Graphite |
|---|---|---|---|---|---|---|---|---|---|
| wt.% | 16.1 | 17.7 | 22.45 | 3.01 | 2.16 | 1.68 | 2.53 | 0.09 | 0.58 |
| Element | Mixture 1 | Mixture 2 | Mixture 3 |
|---|---|---|---|
| Cu-slag + 0 wt.% SBs | Cu-slag + 10 wt.% SBs | Cu-slag + 20 wt.% SBs | |
| Al as Al2O3 | 2.14 | 1.93 | 1.71 |
| Al metallic | - | 1.61 | 3.22 |
| As | 0.19 | 0.17 | 0.15 |
| C 1 | - | 0.058 | 0.116 |
| Ca | 0.805 | 0.72 | 0.64 |
| Co | 0.062 | 1.83 | 3.59 |
| Cu | 1.19 | 2.84 | 5.44 |
| Fe | 38.3 | 34.8 | 31.2 |
| K | 0.89 | 0.80 | 0.71 |
| Li | - | 0.22 | 0.43 |
| Mg | 1.19 | 1.07 | 0.95 |
| Mn | - | 0.17 | 0.34 |
| Ni | 0.048 | 0.30 | 0.54 |
| Pb | 0.31 | 0.28 | 0.25 |
| S | 0.47 | 0.42 | 0.38 |
| Sb | 0.1 | 0.09 | 0.08 |
| Si | 15.4 | 13.9 | 12.3 |
| Zn | 1.78 | 1.60 | 1.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinne, T.; Klemettinen, A.; Klemettinen, L.; Ruismäki, R.; O’Brien, H.; Jokilaakso, A.; Serna-Guerrero, R. Recovering Value from End-of-Life Batteries by Integrating Froth Flotation and Pyrometallurgical Copper-Slag Cleaning. Metals 2022, 12, 15. https://doi.org/10.3390/met12010015
Rinne T, Klemettinen A, Klemettinen L, Ruismäki R, O’Brien H, Jokilaakso A, Serna-Guerrero R. Recovering Value from End-of-Life Batteries by Integrating Froth Flotation and Pyrometallurgical Copper-Slag Cleaning. Metals. 2022; 12(1):15. https://doi.org/10.3390/met12010015
Chicago/Turabian StyleRinne, Tommi, Anna Klemettinen, Lassi Klemettinen, Ronja Ruismäki, Hugh O’Brien, Ari Jokilaakso, and Rodrigo Serna-Guerrero. 2022. "Recovering Value from End-of-Life Batteries by Integrating Froth Flotation and Pyrometallurgical Copper-Slag Cleaning" Metals 12, no. 1: 15. https://doi.org/10.3390/met12010015
APA StyleRinne, T., Klemettinen, A., Klemettinen, L., Ruismäki, R., O’Brien, H., Jokilaakso, A., & Serna-Guerrero, R. (2022). Recovering Value from End-of-Life Batteries by Integrating Froth Flotation and Pyrometallurgical Copper-Slag Cleaning. Metals, 12(1), 15. https://doi.org/10.3390/met12010015