
Local Lattice Distortion in High-Entropy Carbide Ceramics


Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, J.; Zhang, J.; Zhang, F.; Niu, B.; Lei, L.; Wang, W. High-entropy carbide: A novel class of multicomponent ceramics. Ceram. Int. 2018, 44, 22014–22018. [Google Scholar] [CrossRef]
- Nisar, A.; Zhang, C.; Boesl, B.; Agarwal, A. A perspective on challenges and opportunities in developing high entropy-ultra high temperature ceramics. Ceram. Int. 2020, 46, 25845–25853. [Google Scholar] [CrossRef]
- Xiang, H.; Xing, Y.; Dai, F.; Wang, H.; Su, L.; Wang, H.; Zhao, B.; Li, J.; Zhou, Y. High-entropy ceramics: Present status, challenges, and a look forward. J. Adv. Ceram. 2021, 10, 385–441. [Google Scholar] [CrossRef]
- Oses, C.; Toher, C.; Curtarolo, S. High-entropy ceramics. Nat. Rev. Mater. 2020, 5, 295–309. [Google Scholar] [CrossRef]
- Alvi, S.A.; Zhang, H.; Akhtar, F. High-Entropy Ceramics. In Engineering Steels and High Entropy-Alloys; IntechOpen: London, UK, 2019. [Google Scholar]
- Wang, Z.; Li, Z.T.; Zhao, S.J.; Wu, Z.G. High-entropy carbide ceramics: A perspective review. Tungsten 2021, 3, 131–142. [Google Scholar] [CrossRef]
- Lei, Z.; Liu, X.; Wang, H.; Wu, Y.; Jiang, S.; Lu, Z. Development of advanced materials via entropy engineering. Scr. Mater. 2019, 165, 164–169. [Google Scholar] [CrossRef]
- Cantor, B.; Chang, I.T.H.; Knight, P.; Vincent, A.J.B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375, 213–218. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Rost, C.M.; Sachet, E.; Borman, T.; Moballegh, A.; Dickey, E.C.; Hou, D.; Jones, J.L.; Curtarolo, S.; Maria, J. Entropy-stabilized oxides. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gild, J.; Zhang, Y.; Harrington, T.; Jiang, S.; Hu, T.; Quinn, M.C.; Mellor, W.M.; Zhou, N.; Vecchio, K.; Luo, J. High-Entropy Metal Diborides: A New Class of High-Entropy Materials and a New Type of Ultrahigh Temperature Ceramics. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Sarker, P.; Harrington, T.; Toher, C.; Oses, C.; Samiee, M.; Maria, J.P.; Brenner, D.W.; Vecchio, K.S.; Curtarolo, S. High-entropy high-hardness metal carbides discovered by entropy descriptors. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.F.; Liu, J.X.; Li, F.; Qin, Y.; Liang, Y.C.; Zhang, G.J. High entropy carbide ceramics from different starting materials. J. Eur. Ceram. Soc. 2019, 39, 2989–2994. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, A.; Jia, J.; Meng, J.; Su, B. Phase evolution and properties of (VNbTaMoW) C high entropy carbide prepared by reaction synthesis. J. Eur. Ceram. Soc. 2020, 40, 2746–2751. [Google Scholar] [CrossRef]
- Dai, F.Z.; Wen, B.; Sun, Y.; Xiang, H.; Zhou, Y. Theoretical prediction on thermal and mechanical properties of high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C by deep learning potential. J. Mater. Sci. Technol. 2020, 43, 168–174. [Google Scholar] [CrossRef]
- Ye, B.; Wen, T.; Huang, K.; Wang, C.Z.; Chu, Y. First-principles study, fabrication, and characterization of (Hf0.2Zr0.2Ta0.2Nb0.2Ti0.2 )C high-entropy ceramic. J. Am. Ceram. Soc. 2019, 102, 4344–4352. [Google Scholar] [CrossRef] [Green Version]
- Harrington, T.J.; Gild, J.; Sarker, P.; Toher, C.; Rost, C.M.; Dippo, O.F.; McElfresh, C.; Kaufmann, K.; Marin, E.; Borowski, L.; et al. Phase stability and mechanical properties of novel high entropy transition metal carbides. Acta Mater. 2019, 166, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Dippo, O.F.; Vecchio, K.S. A universal configurational entropy metric for high-entropy materials. Scr. Mater. 2021, 201, 113974. [Google Scholar] [CrossRef]
- Ye, B.; Chu, Y.; Huang, K.; Liu, D. Synthesis and characterization of (Zr1/3Nb1/3Ti1/3)C metal carbide solid-solution ceramic. J. Am. Ceram. Soc. 2019, 102, 919–923. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Z.; Wu, Z.; Xu, B.; Zhao, S.; Zhang, W.; Lin, N. Phase, microstructure and related mechanical properties of a series of (NbTaZr)C-Based high entropy ceramics. Ceram. Int. 2021, 47, 14341–14347. [Google Scholar] [CrossRef]
- Castle, E.; Csanádi, T.; Grasso, S.; Dusza, J.; Reece, M. Processing and Properties of High-Entropy Ultra-High Temperature Carbides. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S. Lattice distortion in high-entropy carbide ceramics from first-principles calculations. J. Am. Ceram. Soc. 2021, 104, 1874–1886. [Google Scholar] [CrossRef]
- Song, H.; Tian, F.; Hu, Q.; Vitos, L.; Wang, Y.; Shen, J.; Chen, N. Local lattice distortion in high-entropy alloys. Phys. Rev. Mater. 2017, 1, 023404. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Lin, D.Y.; Gao, X.; Zhao, Y.F.; Song, H.F. A structural modeling approach to solid solutions based on the similar atomic environment. J. Chem. Phys. 2020, 153, 034101. [Google Scholar] [CrossRef]
- Guan, H.; Huang, S.; Ding, J.; Tian, F.; Xu, Q.; Zhao, J. Chemical environment and magnetic moment effects on point defect formations in CoCrNi-based concentrated solid-solution alloys. Acta Mater. 2020, 187, 122–134. [Google Scholar] [CrossRef]
- Tian, F.; Yang, Z.; Lin, D.Y.; Zhao, Y.F. Lattice distortion inducing local antiferromagnetic behaviors in FeAl alloys. J. Phys. Condens. Matter 2020, 32. [Google Scholar] [CrossRef]
- Kresse, G.; Marsman, M.; Furthmüller, J. Vienna Ab-initio Package Vienna Simulation: VASP the GUIDE; VASP Software GmbH: Vienna, Austria, 2016. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Tian, F.; Varga, L.K.; Chen, N.; Shen, J.; Vitos, L. Empirical design of single phase high-entropy alloys with high hardness. Intermetallics 2015, 58, 1–6. [Google Scholar] [CrossRef]
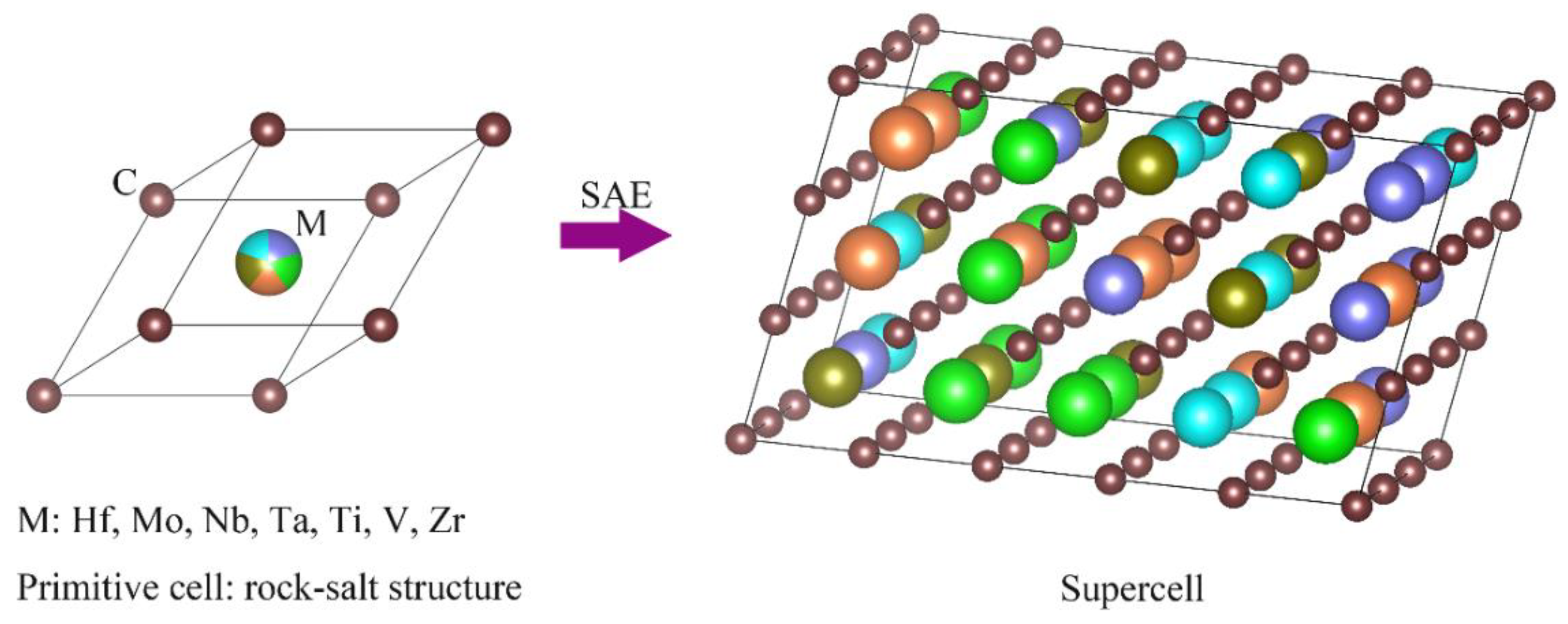
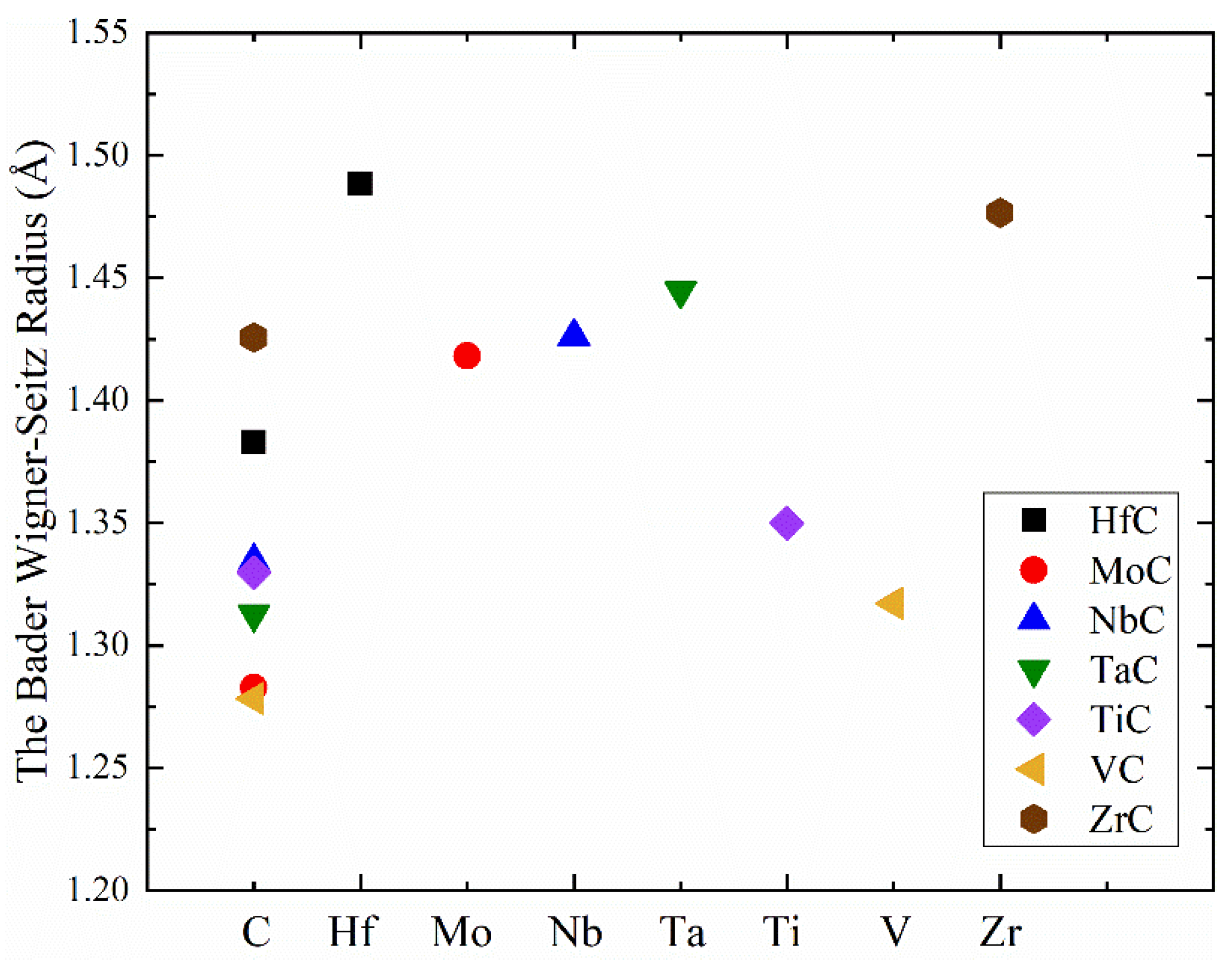



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HECs | ΔH1 | ΔH2 | Δd | δ | Exp. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | Hf | Mo | Nb | Ta | Ti | V | Zr | tot. | |||||
| HfNbTaTiVC5 | 1.330 | −64.85 | 0.062 | 0.045 | -- | 0.065 | 0.039 | 0.050 | 0.084 | -- | 0.059 | 4.514 | 0.107 |
| HfNbTaTiZrC5 | −0.902 | −74.82 | 0.062 | 0.039 | -- | 0.033 | 0.038 | 0.027 | -- | 0.037 | 0.048 | 3.884 | 0.094 |
| MoNbTaTiVC5 | −1.276 | −47.41 | 0.049 | -- | 0.102 | 0.046 | 0.044 | 0.043 | 0.105 | -- | 0.060 | 4.223 | -- |
| HECs | L | Exp. | LLD | Ideal | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | a | b | c | |||
| HfNbTaTiVC5 | 4.432 | 4.415 | 9.403 | 9.402 | 15.677 | 59.923 | 59.966 | 60.012 | 9.420 | 9.420 | 15.700 |
| HfNbTaTiZrC5 | 4.519 | 4.500 | 9.589 | 9.591 | 15.973 | 59.971 | 59.982 | 59.985 | 9.585 | 9.585 | 15.975 |
| MoNbTaTiVC5 | 4.375 | -- | 9.282 | 9.278 | 15.461 | 59.990 | 60.071 | 59.992 | 9.285 | 9.285 | 15.475 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, H.; Cui, C.; Song, H.; Tian, F. Local Lattice Distortion in High-Entropy Carbide Ceramics. Metals 2021, 11, 1399. https://doi.org/10.3390/met11091399
Ge H, Cui C, Song H, Tian F. Local Lattice Distortion in High-Entropy Carbide Ceramics. Metals. 2021; 11(9):1399. https://doi.org/10.3390/met11091399
Chicago/Turabian StyleGe, Huijuan, Chengfeng Cui, Hongquan Song, and Fuyang Tian. 2021. "Local Lattice Distortion in High-Entropy Carbide Ceramics" Metals 11, no. 9: 1399. https://doi.org/10.3390/met11091399
APA StyleGe, H., Cui, C., Song, H., & Tian, F. (2021). Local Lattice Distortion in High-Entropy Carbide Ceramics. Metals, 11(9), 1399. https://doi.org/10.3390/met11091399