Calculating Study on Properties of Al (111)/6H-SiC (0001) Interfaces


Abstract
1. Introduction
2. Details of Calculation Methods
3. Results and Discussion
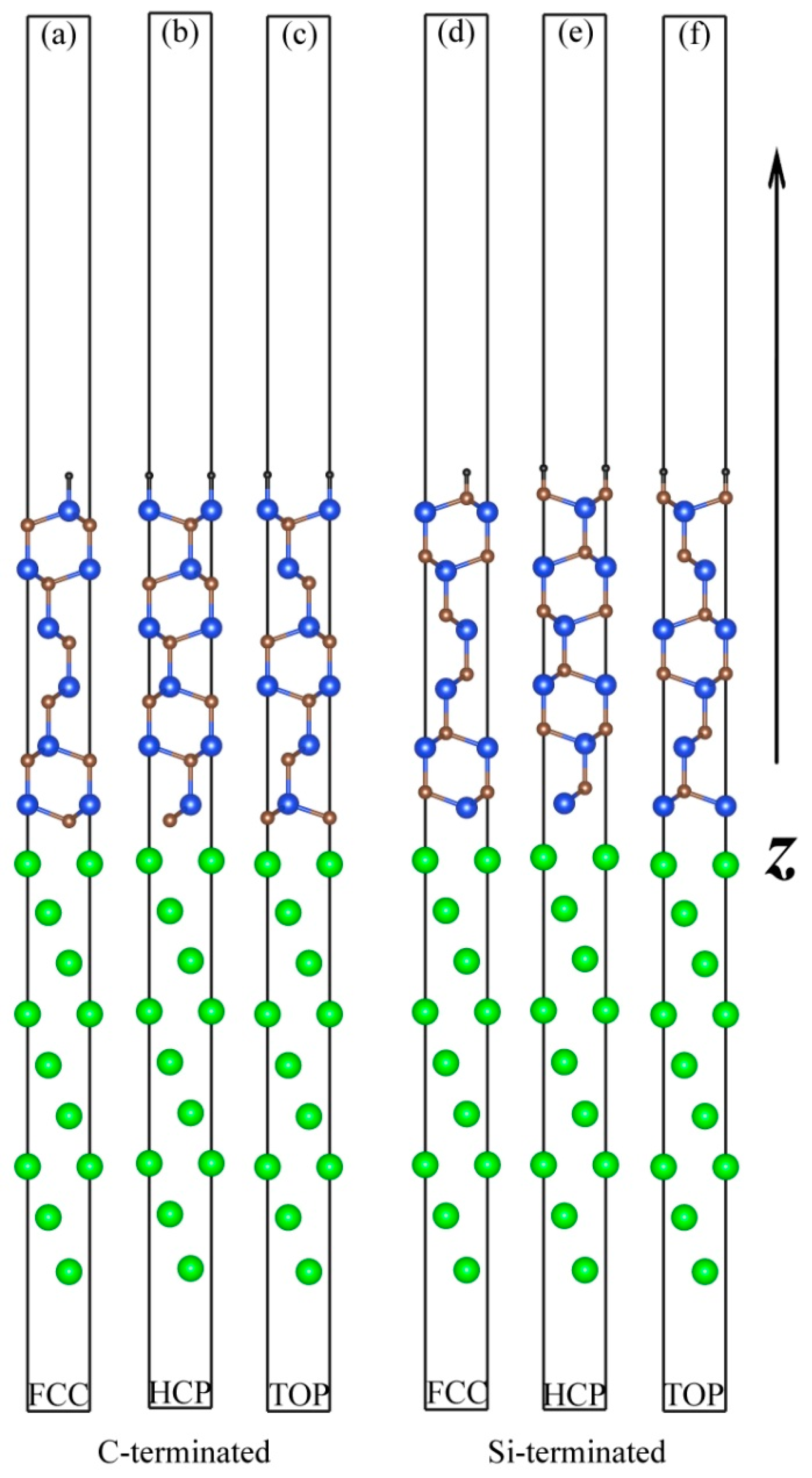
3.1. Atomic Structures of the Al (111)/6H-SiC (0001) Interfaces
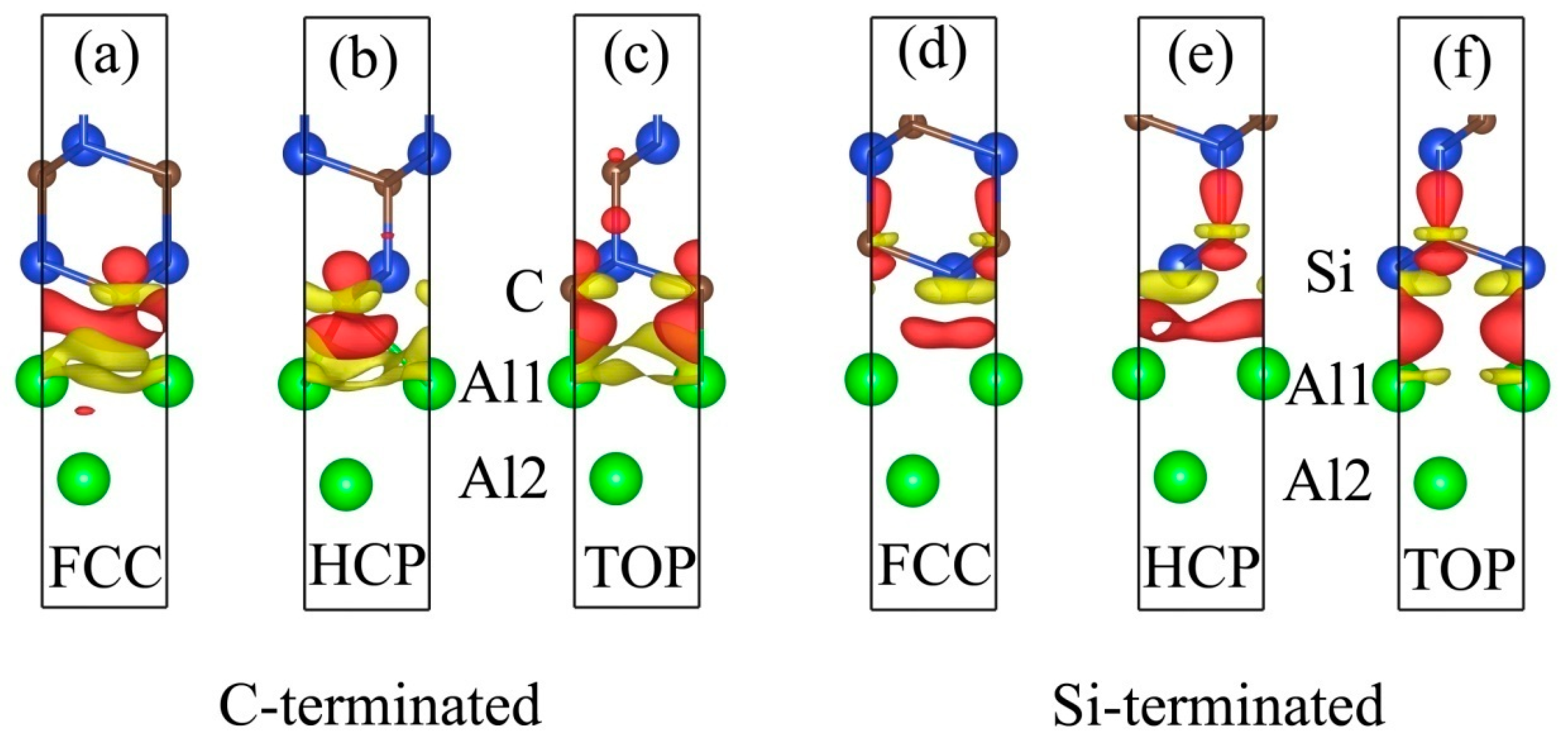
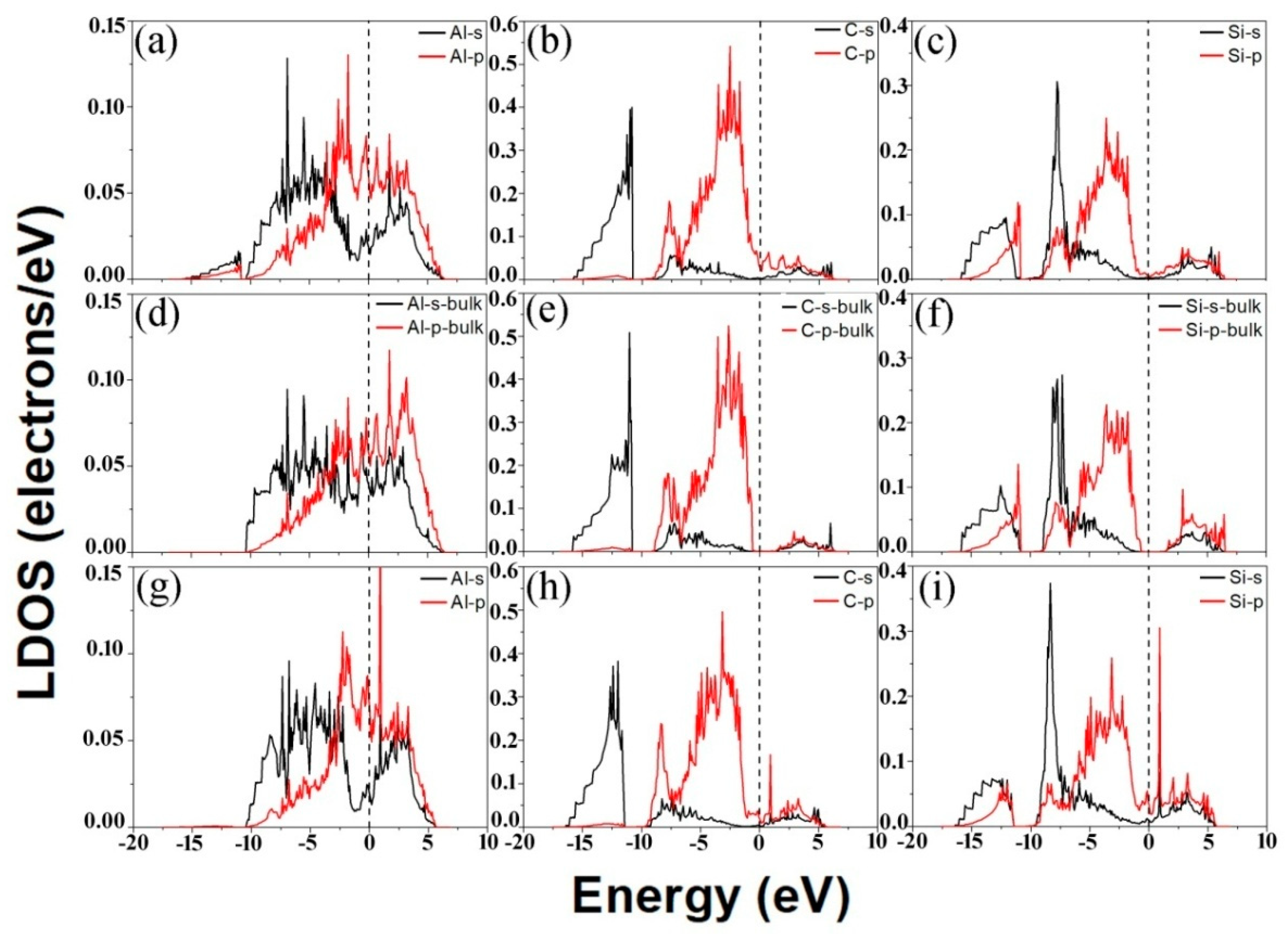
3.2. Electronic Structures of the Al (111)/6H-SiC (0001) Interfaces
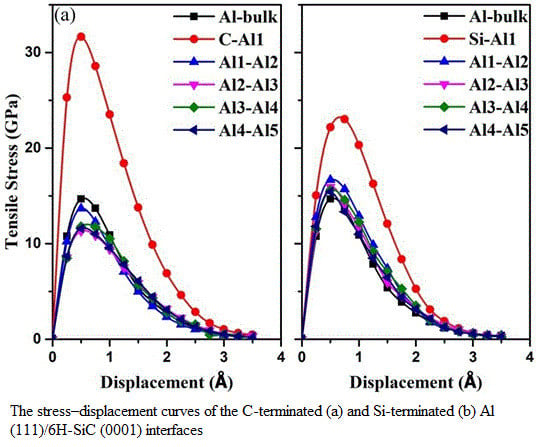
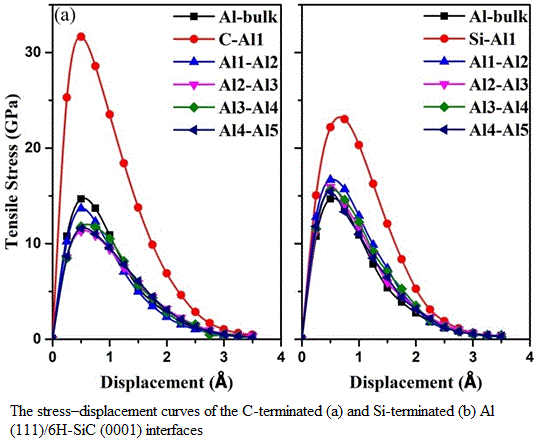
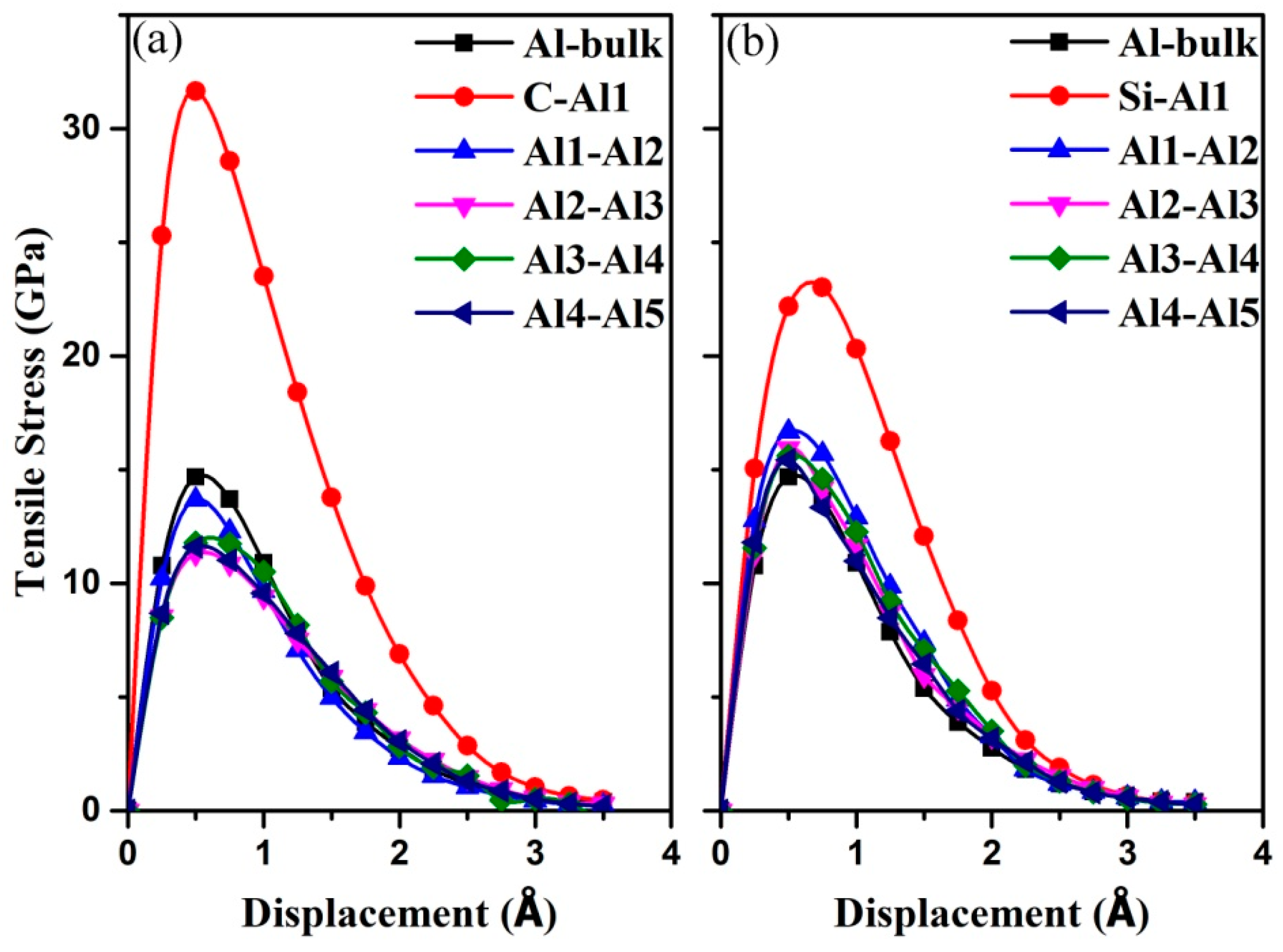
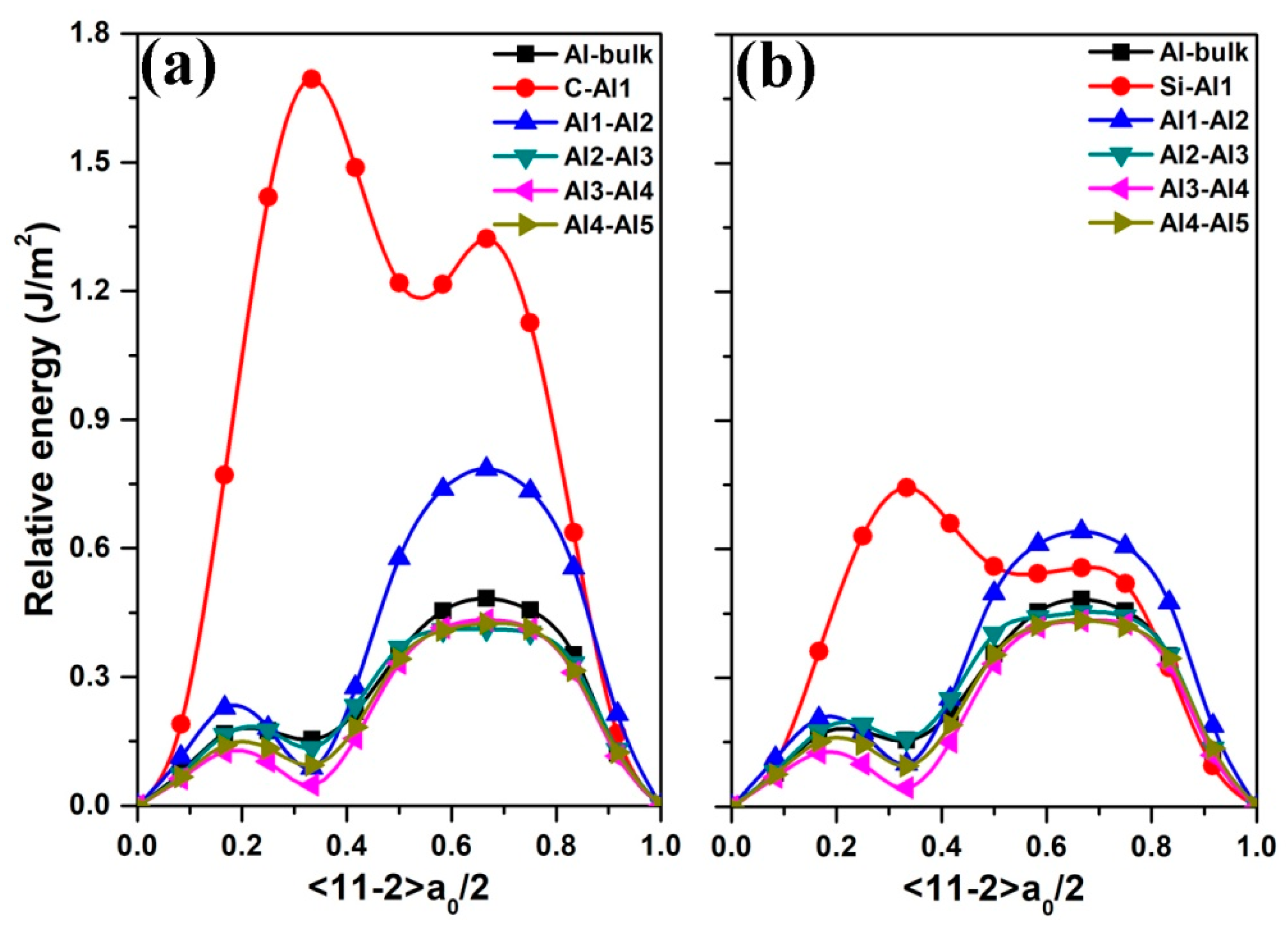
3.3. Mechanical Properties of the Al (111)/6H-SiC (0001) Interfaces
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Casati, R.; Vedani, M. Metal Matrix Composites Reinforced by Nano-Particles-A Review. Metals 2014, 4, 65–83. [Google Scholar] [CrossRef]
- Saheb, N.; Qadir, N.U.; Siddiqui, M.U.; Arif, A.F.M.; Akhtar, S.S.; Aqeeli, N.A. Characterization of Nano reinforcement Dispersion in Inorganic Nano composites: A Review. Materials 2014, 7, 4148–4181. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Mishra, P.C.; Singh, S.; Thakur, R.K. Tool wear in turning ceramic reinforced aluminum matrix composites-A review. J. Compos. Mater. 2015, 49, 2949–2961. [Google Scholar]
- Singh, J.; Chauhan, A. Overview of wear performance of aluminium matrix composites reinforced with ceramic materials under the influence of controllable variables. Ceram. Int. 2016, 42, 56–81. [Google Scholar] [CrossRef]
- Kim, C.S.; Cho, K.; Manjili, M.H.; Nezafati, M. Mechanical performance of particulate-reinforced Al metal-matrix composites (MMCs) and Al metal-matrix nano-composites (MMNCs). J. Mater. Sci. 2017, 52, 13319–13349. [Google Scholar] [CrossRef]
- Wunderlich, W. The Atomistic Structure of Metal/Ceramic Interfaces Is the Key Issue for Developing Better Properties. Metals 2014, 4, 410–427. [Google Scholar] [CrossRef]
- Li, N.; Liu, X.Y. Review: Mechanical behavior of metal/ceramic interfaces in nano layered composites-experiments and modeling. J. Mater. Sci. 2018, 53, 5562–5583. [Google Scholar] [CrossRef]
- Li, S.; Arsenault, R.J.; Jena, P. Quantum chemical study of adhesion at the SiC/Al interface. J. Appl. Phys. 1988, 64, 6246–6253. [Google Scholar] [CrossRef]
- Aboelfotoh, M.O.; Frojdh, C.; Petersson, C.S. Schottky-barrier behavior of metals on n- and p-type 6H-SiC. Phys. Rev. B 2003, 67, 075312. [Google Scholar] [CrossRef]
- Tanaka, S.; Tamura, T.; Okazaki, K.; Ishibashi, S.; Kohyama, M. First-Principles Calculations of Schottky Barrier Heightsof Monolayer Metal/6H-SiC{0001} Interfaces. Mater. Trans. 2006, 47, 2690–2695. [Google Scholar] [CrossRef]
- Tanaka, S.; Tamura, T.; Okazaki, K.; Ishibashi, S.; Kohyama, M. Schottky-barrier heights of metal/alpha-SiC{0001} interfaces by first-principles calculations. Phys. Status Solidi C 2007, 4, 2972–2976. [Google Scholar] [CrossRef]
- Xu, X.Y.; Wang, H.Y.; Zha, M.; Wang, C.; Yang, Z.Z.; Jiang, Q.C. Effects of Ti, Si, Mg and Cu additions on interfacial properties and electronic structure of Al(111)/4H-SiC(0001) interface: A first-principles study. Appl. Surf. Sci. 2018, 437, 103–109. [Google Scholar] [CrossRef]
- Wu, Q.; Xie, J.; Wang, A.; Wang, C.; Mao, A. Effects of vacancies at Al(111)/6H-SiC(0001) interfaces on deformation behavior: A first-principle study. Comput. Mater. Sci. 2019, 158, 110–116. [Google Scholar] [CrossRef]
- Wu, Q.; Xie, J.; Wang, C.; Li, L.; Wang, A.; Mao, A. First-principles study of the structure properties of Al(111)/6H-SiC(0001) interfaces. Surf. Sci. 2018, 670, 1–7. [Google Scholar] [CrossRef]
- Wang, C.Q.; Chang, D.H.; Jia, Y.; Xie, J.P. Electronic and mechanical properties of Al (100)/6H–SiC (0001) interfaces: A first-principles study. Mater. Res. Express 2019, 6, 126316. [Google Scholar] [CrossRef]
- Liu, P.; Wang, A.; Xie, J.; Hao, S. Characterization and evaluation of interface in SiCp/2024 Al composite. Trans. Nonferrous Met. Soc. Chin. 2015, 25, 1410–1418. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmiiller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillonin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Dudiy, S.V.; Lundqvist, B.I. Wetting of TiC and TiN by metals. Phys. Rev. B 2004, 69, 125421. [Google Scholar] [CrossRef]
- Xie, C.; Xu, P.; Xu, F.; Pan, H.; Li, Y. First-principles studies of the electronic and optical properties of 6H–SiC. Phys. B 2003, 336, 284–289. [Google Scholar] [CrossRef]
- Yadav, S.K.; Ramprasad, R.; Misra, A.; Liu, X.Y. Core structure and Peierls stress of edge and screw dislocations in TiN: A density functional theory study. Acta. Mater. 2014, 74, 268–277. [Google Scholar] [CrossRef]
- Qi, Y.; Mishra, R.K. Ab initio study of the effect of solute atoms on the stacking fault energy in aluminum. Phys. Rev. B 2007, 75, 224105. [Google Scholar] [CrossRef]
- Hirth, J.P.; Lothe, J. Theory of Dislocations, 2nd ed.; Wiley: Hoboken, NJ, USA, 1982. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interfaces | d0 (Å) | r (Å) | ||
|---|---|---|---|---|
| C-terminated | TOP | 1.99 | 1.99 | 3.90 |
| HCP | 1.74 | 2.49 | 2.67 | |
| FCC | 1.83 | 2.56 | 2.27 | |
| Si-terminated | TOP | 2.53 | 2.53 | 2.93 |
| HCP | 2.32 | 2.92 | 2.29 | |
| FCC | 2.25 | 2.88 | 2.15 | |
| Interfaces | d0 (Å) | (GPa) | (J/m2) | |
|---|---|---|---|---|
| C-terminated | C-Al1 | 1.99 | 31.65 | 1.32 |
| Al1-Al2 | 2.08 | 13.68 | 0.23 | |
| Al2-Al3 | 2.18 | 11.30 | 0.18 | |
| Al3-Al4 | 2.20 | 11.78 | 0.12 | |
| Al4-Al5 | 2.32 | 11.59 | 0.14 | |
| Si-terminatedbulk | Si-Al1 | 2.53 | 22.18 | 0.56 |
| Al1-Al2 | 2.09 | 16.68 | 0.21 | |
| Al2-Al3 | 2.17 | 15.94 | 0.19 | |
| Al3-Al4 | 2.25 | 15.61 | 0.13 | |
| Al4-Al5 | 2.32 | 15.44 | 0.15 | |
| - | 2.33 | 14.68 | 0.17 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Chen, W.; Jia, Y.; Xie, J. Calculating Study on Properties of Al (111)/6H-SiC (0001) Interfaces. Metals 2020, 10, 1197. https://doi.org/10.3390/met10091197
Wang C, Chen W, Jia Y, Xie J. Calculating Study on Properties of Al (111)/6H-SiC (0001) Interfaces. Metals. 2020; 10(9):1197. https://doi.org/10.3390/met10091197
Chicago/Turabian StyleWang, Changqing, Weiguang Chen, Yu Jia, and Jingpei Xie. 2020. "Calculating Study on Properties of Al (111)/6H-SiC (0001) Interfaces" Metals 10, no. 9: 1197. https://doi.org/10.3390/met10091197
APA StyleWang, C., Chen, W., Jia, Y., & Xie, J. (2020). Calculating Study on Properties of Al (111)/6H-SiC (0001) Interfaces. Metals, 10(9), 1197. https://doi.org/10.3390/met10091197