DFT-CEF Approach for the Thermodynamic Properties and Volume of Stable and Metastable Al–Ni Compounds

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Methodology
2.1. DFT Calculations
2.2. Thermodynamic Modeling
3. Results
3.1. Structural Stability at 0 K for Al And Ni
3.2. Phase Stability and Molar Volumes of Binary Phases at 0 K
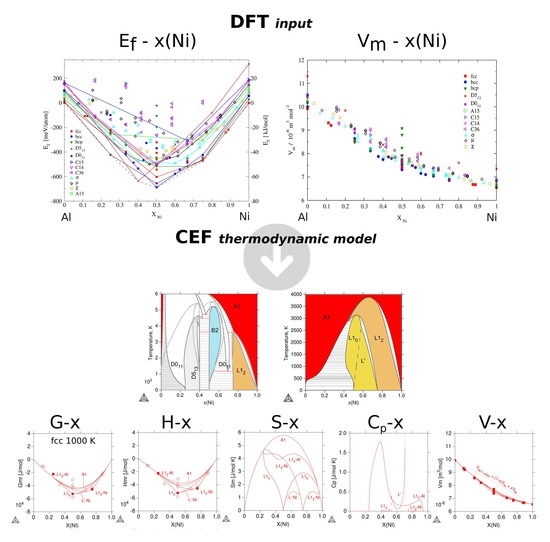
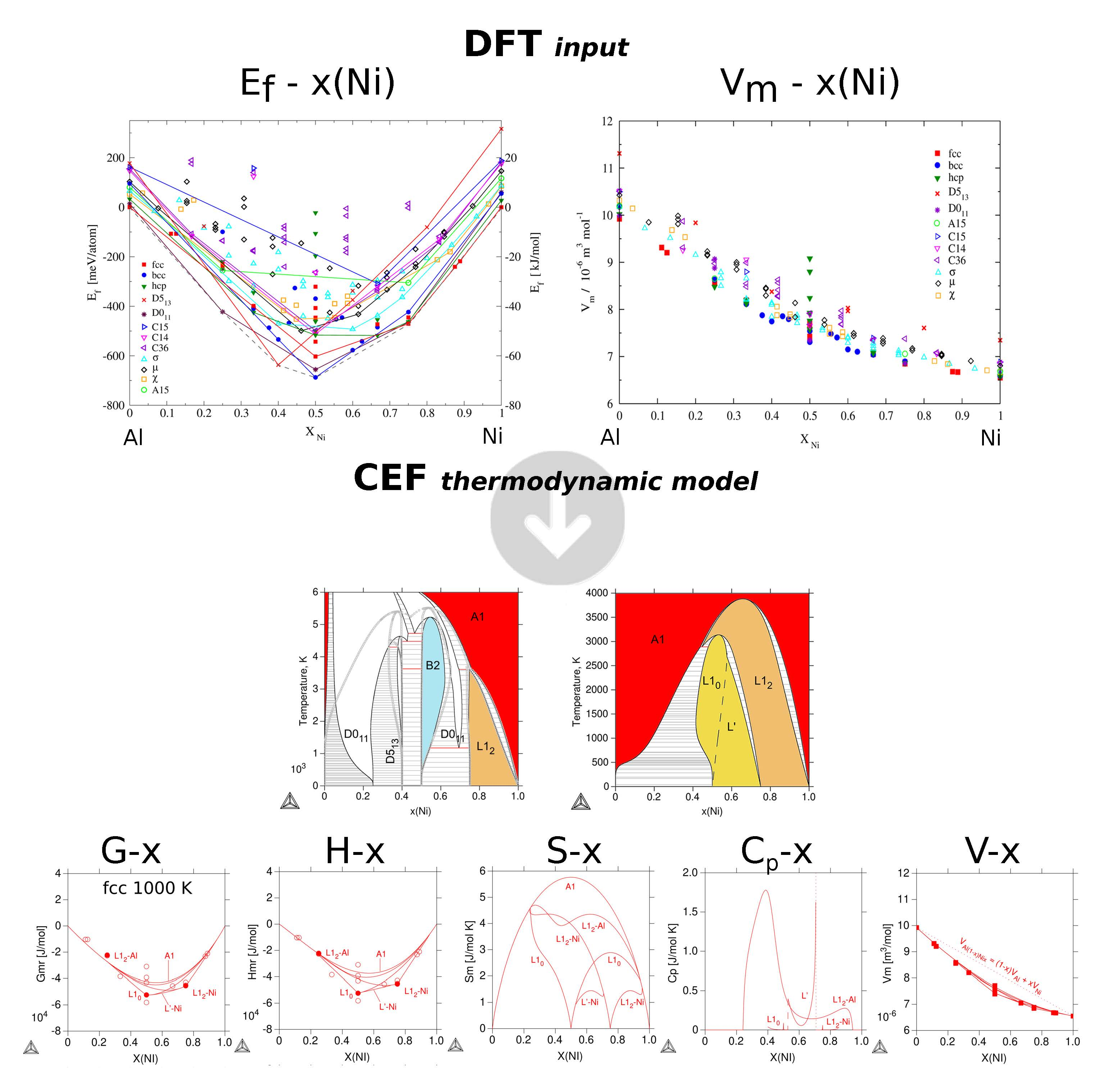
3.2.1. Enthalpies of Formation of Ordered Compounds
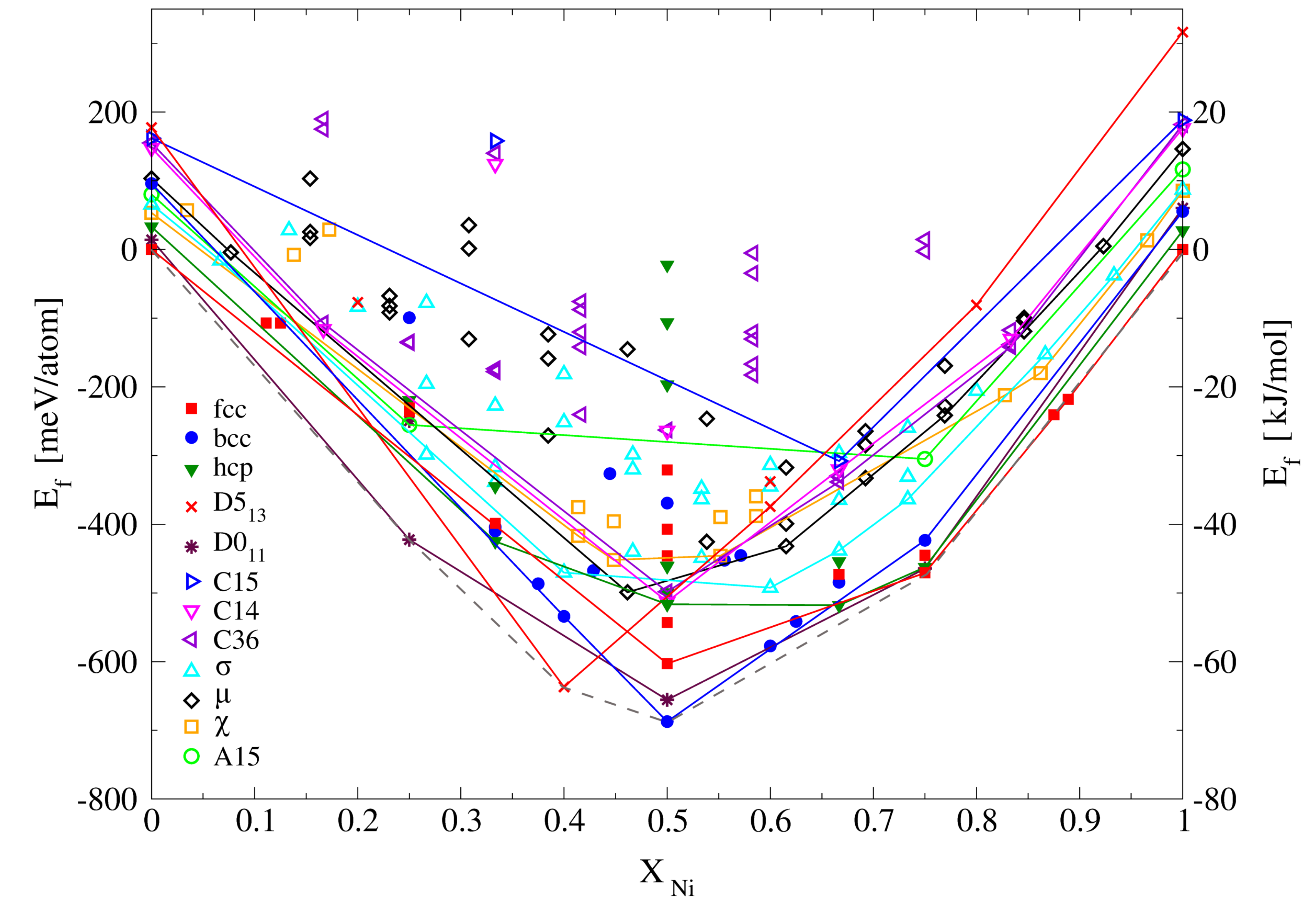
3.2.2. Molar Volumes of Ordered Compounds
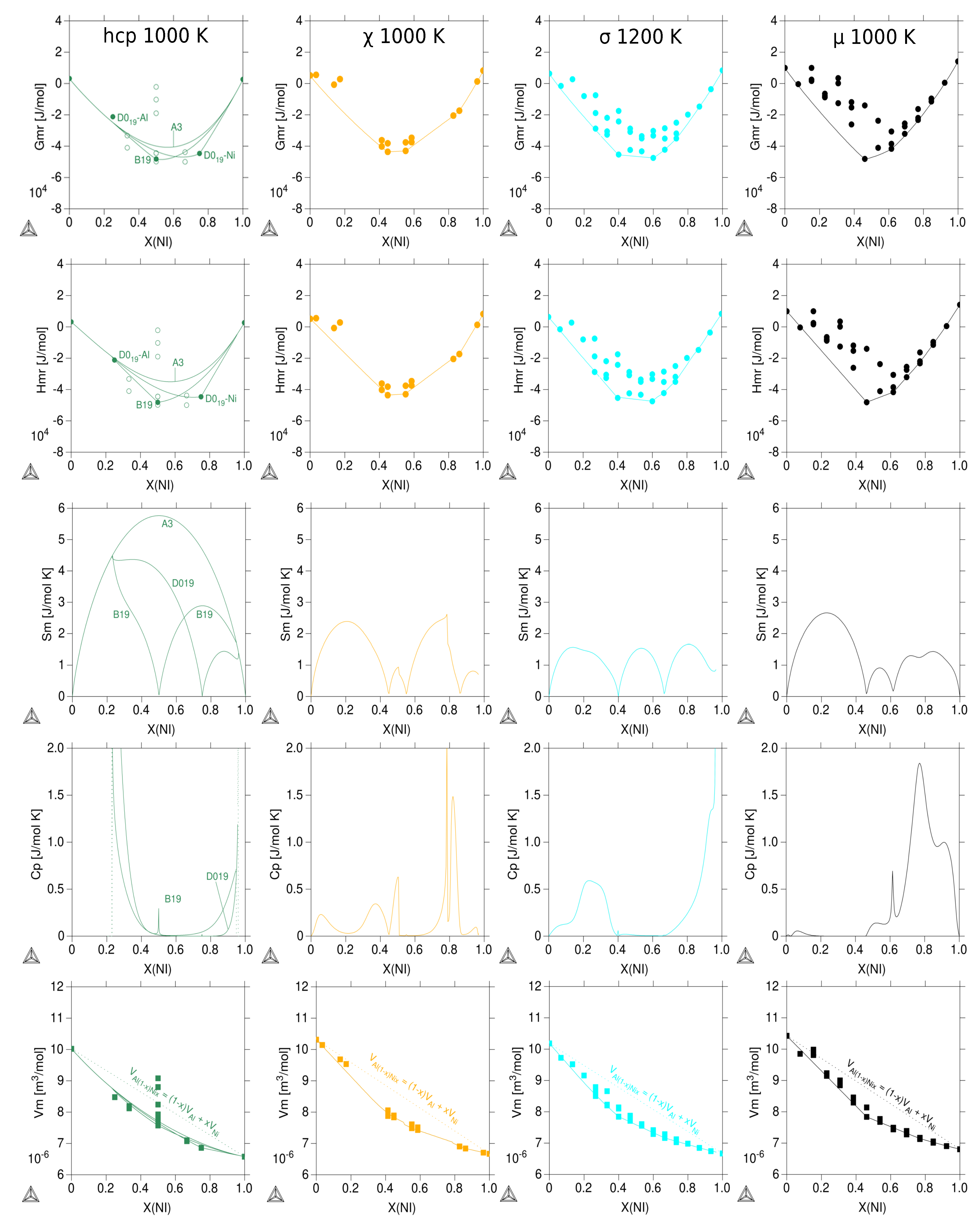
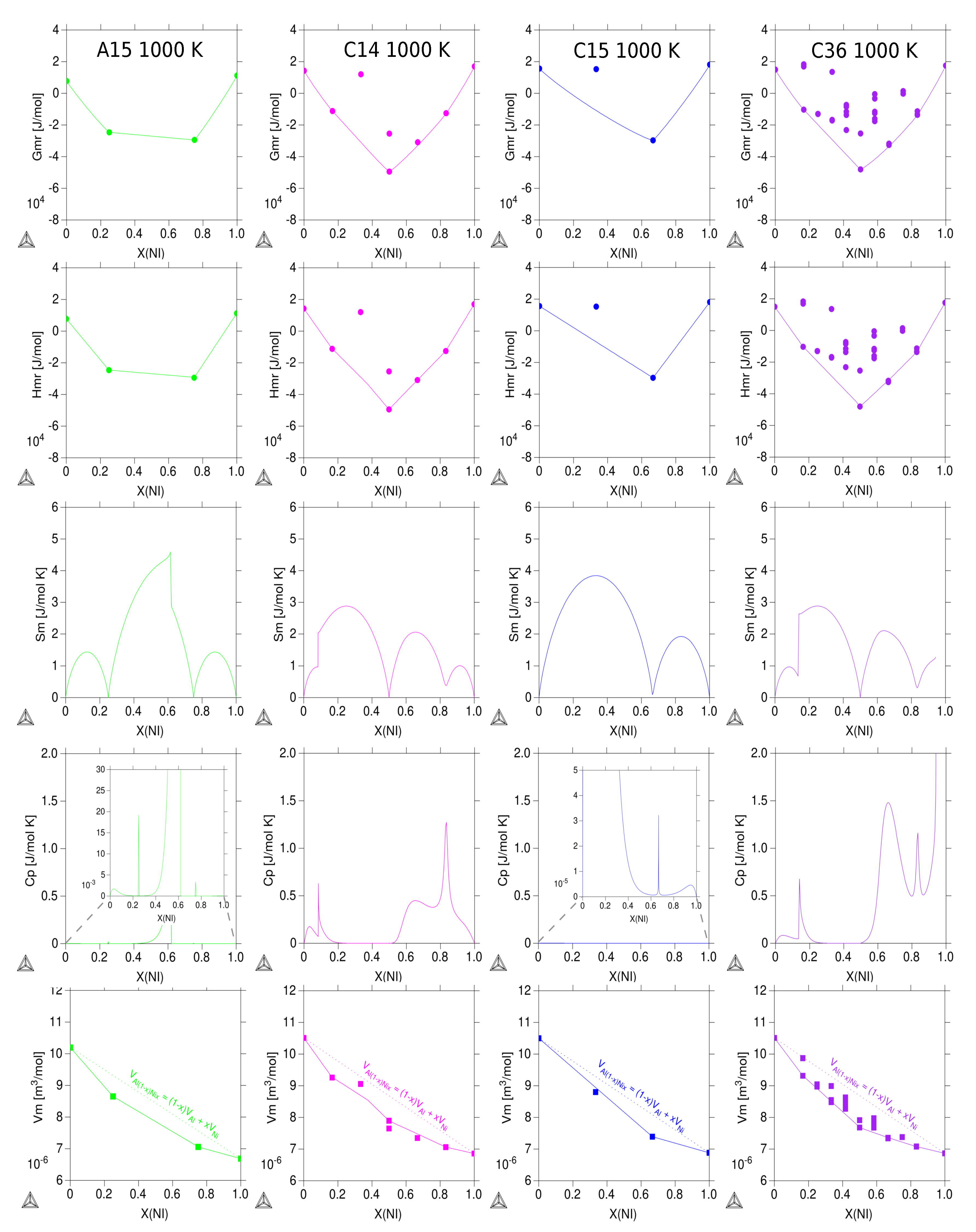
3.3. Thermodynamic Properties of Individual Phases at Finite Temperature
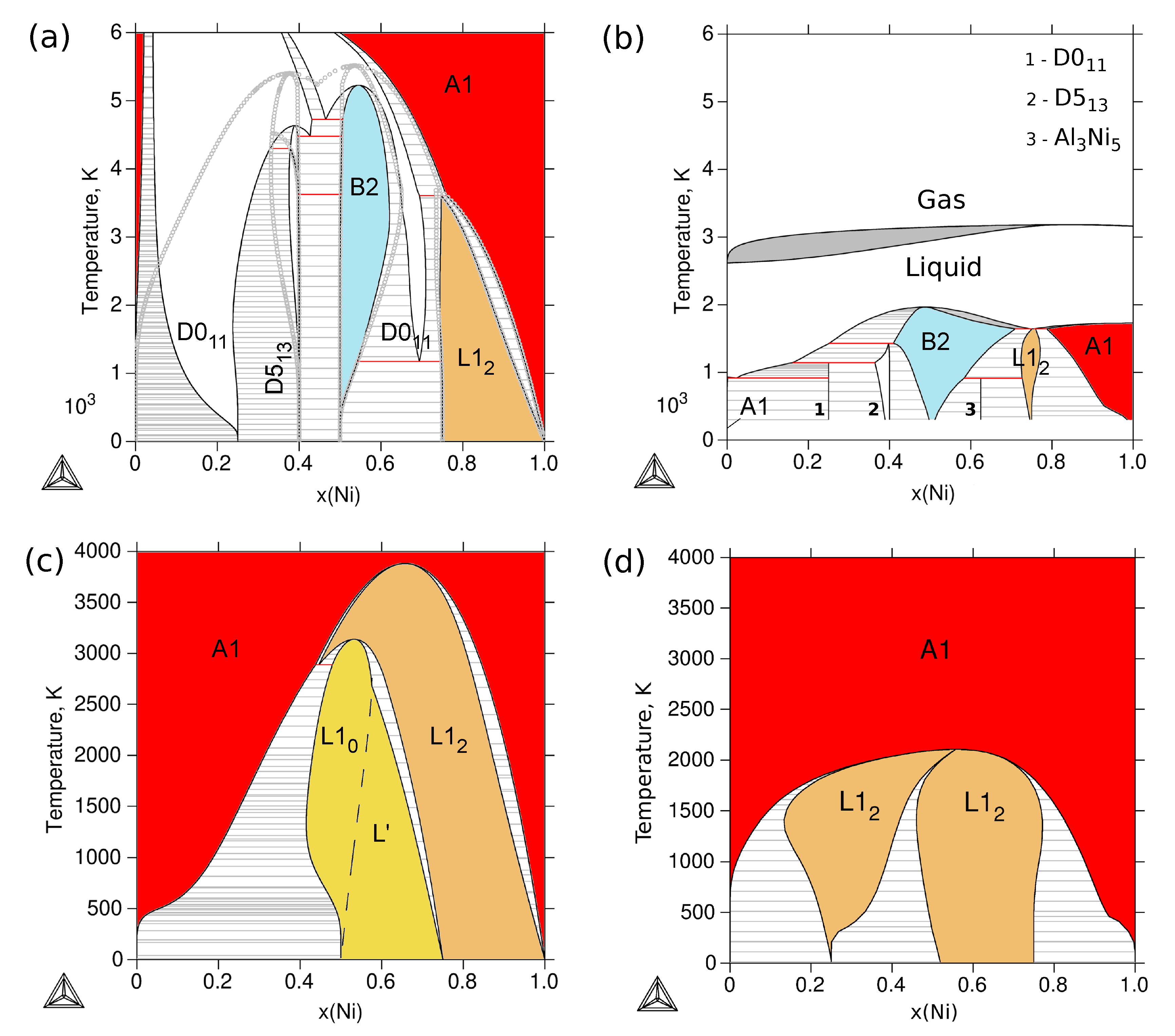
3.4. Phase Stability at Finite Temperature—The Phase Diagram
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATAT | Alloy Theoretic Automated Toolkit |
| BWG | Bragg–Williams–Gorsky |
| Calphad | CALculation of PHAse Diagrams |
| CE | Cluster Expansion |
| CEF | Compound Energy Formalism |
| CVM | Cluster Variation Method |
| DFT | Density Functional Theory |
| FP | First Principles |
| FPLMTO | Full-Potential Linear Muffin-Tin Orbital |
| GGA | Generalized Gradient Approximation |
| LMTO | Linear Muffin-Tin Orbital |
| MC | Monte Carlo Simulations |
| MD | Molecular Dynamics |
| PAW | Projector Augmented Wave |
| PBE | Perdew-Burke-Ernzerhof |
| SL | Sublattice(-s) |
| SGTE | Scientific Group Thermodata Europe |
| TCP | Topologically Closed Packed |
| TDB | Thermodynamic Database |
| VASP | Vienna Ab initio Simulation Package |
References
- Fries, S.G.; Lukas, H.L.; Ansara, I.; Sundman, B. The Bragg-Williams-Gorsky (BWG) ordering treatment in the compound energy formalism (CEF). Berichte Der Bunsenges. Für Phys. Chem. 1998, 102, 1102–1110. [Google Scholar] [CrossRef]
- Lukas, H.; Fries, S.G.; Sundman, B. Computational Thermodynamics. The Calphad Method; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Martin, R. Electronic Structure. Basic Theory and Practical Methods; Cambridge University Press: Urbana, IL, USA; Champaign, IL, USA, 2004. [Google Scholar]
- Dupin, N.; Kattner, U.R.; Sundman, B.; Palumbo, M.; Fries, S.G. Implementation of an Effective Bond Energy Formalism in the Multicomponent Calphad Approach. J. Res. Natl. Inst. Stand. Technol. 2018, 123, 123020. [Google Scholar] [CrossRef]
- Reed, R.C. The Superalloys; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Dupin, N.; Sundman, B. A thermodynamic database for Ni-base superalloys. Scand. J. Metall. 2001, 30, 184–192. [Google Scholar] [CrossRef]
- Parsa, A.B.; Wollgramm, P.; Buck, H.; Somsen, C.; Kostka, A.; Povstugar, I.; Choi, P.P.; Raabe, D.; Dlouhy, A.; Müller, J.; et al. Advanced scale bridging microstructure analysis of single crystal Ni-base superalloys. Adv. Eng. Mater. 2015, 17, 216–230. [Google Scholar] [CrossRef]
- Lopez-Galilea, I.; Koßmann, J.; Kostka, A.; Drautz, R.; Mujica Roncery, L.; Hammerschmidt, T.; Huth, S.; Theisen, W. The thermal stability of topologically close-packed phases in the single-crystal Ni-base superalloy ERBO/1. J. Mater. Sci. 2016, 51, 2653–2664. [Google Scholar] [CrossRef]
- Goiri, J.G.; Van Der Ven, A. Phase and structural stability in Ni-Al systems from first principles. Phys. Rev. B 2016, 94, 16–19. [Google Scholar] [CrossRef]
- Palumbo, M.; Fries, S.G.; Hammerschmidt, T.; Abe, T.; Crivello, J.C.; Breidi, A.A.H.; Joubert, J.M.; Drautz, R. First-principles-based phase diagrams and thermodynamic properties of TCP phases in Re–X systems (X = Ta, V, W). Comput. Mater. Sci. 2014, 81, 433–445. [Google Scholar] [CrossRef]
- Andersson, J.O.; Helander, T.; Höglund, L.; Shi, P.; Sundman, B. Thermo-Calc & DICTRA, computational tools for materials science. Calphad 2002, 26, 273–312. [Google Scholar]
- Thermo-Calc. Available online: http://www.thermocalc.com (accessed on 17 June 2020).
- van de Walle, A.; Sun, R.; Hong, Q.J.; Kadkhodaei, S. Software tools for high-throughput CALPHAD from first-principles data. Calphad 2017, 58, 70–81. [Google Scholar] [CrossRef]
- Wang, P.; Koßmann, J.; Kattner, U.R.; Palumbo, M.; Hammerschmidt, T.; Olson, G.B. Thermodynamic assessment of the Co-Ta system. Calphad 2019, 64, 205–212. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, T.; Bialon, A.; Pettifor, D.; Drautz, R. Topologically closed-packed phases in binary transition-metal compounds: Matching high-throughput ab-initio calculations to an empirical structure-map. New J. Phys. 2013, 15, 115016. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Fries, S.G.; Sundman, B. Using Re-W σ-phase first-principles results in the Bragg-Williams approximation to calculate finite-temperature thermodynamic properties. Phys. Rev. B 2002, 66, 012203. [Google Scholar] [CrossRef]
- Andersson, J.O.; Guillermet, A.; Hillert, M. A compound-energy model of ordering in a phase with sites of different coordination numbers. Acta Metall. 1986, 34, 437–445. [Google Scholar] [CrossRef]
- Hillert, M. The compound energy formalism. J. Alloys Compd. 2001, 320, 161–176. [Google Scholar] [CrossRef]
- Sluiter, M. Ab initio lattice stabilities of some elemental complex structures. Calphad 2006, 30, 357–366. [Google Scholar] [CrossRef]
- Wang, Y.; Curtarolo, S.; Jiang, C.; Arroyave, R.; Wang, T.; Ceder, G.; Chen, L.Q.; Liu, Z.K. Ab initio lattice stability in comparison with CALPHAD lattice stability. Calphad 2004, 28, 79–90. [Google Scholar] [CrossRef]
- Dinsdale, A.T. SGTE data for pure elements. Calphad 1991, 15, 317–425. [Google Scholar] [CrossRef]
- Palumbo, M. (ICAMS–RUB, Bochum, Germany). Personal communication. 2015–2016.
- Sin’ko, G.V.; Smirnov, N.A. Ab initio calculations of elastic constants and thermodynamic properties of bcc, fcc, and hcp Al crystals under pressure. J. Phys. Condens. Matter 2002, 14, 6989–7005. [Google Scholar]
- Lu, Z.W.; Wei, S.H.H.; Zunger, A.; Frota-Pessoa, S.; Ferreira, L.G. First-principles statistical mechanics of structural stability of intermetallic compounds. Phys. Rev. B 1991, 44, 512–544. [Google Scholar] [CrossRef]
- Yan, J.Y.; Olson, G.B. Molar volumes of bcc, hcp, and orthorhombic Ti-base solid solutions at room temperature. Calphad 2016, 52, 152–158. [Google Scholar] [CrossRef]
- Hallstedt, B. Molar volumes of Al, Li, Mg and Si. Calphad 2007, 31, 292–302. [Google Scholar] [CrossRef]
- Syassen, K.; Holzapfel, W.B. Isothermal compression of Al and Ag to 120 kbar. J. Appl. Phys. 1978, 49, 4427–4430. [Google Scholar] [CrossRef]
- Palumbo, M.; Fries, S.G.; Dal Corso, A.; Körmann, F.; Hickel, T.; Neugebauer, J. Reliability evaluation of thermophysical properties from first-principles calculations. J. Phys. Condens. Matter 2014, 26, 335401. [Google Scholar] [CrossRef]
- Rasamny, M.; Weinert, M.; Fernando, G.W.; Watson, R.E. Electronic structure of a neutral oxygen vacancy in Al3Ni. Phys. Rev. B 2001, 64, 144107. [Google Scholar] [CrossRef]
- Saniz, R.; Ye, L.H.; Shishidou, T.; Freeman, A.J. Structural, electronic, and optical properties of Al3Ni: First-principles calculations. Phys. Rev. B 2006, 74, 014209. [Google Scholar] [CrossRef]
- Chrifi-Alaoui, F.; Nassik, M.; Mahdouk, K.; Gachon, J. Enthalpies of formation of the Al–Ni intermetallic compounds. J. Alloys Compd. 2004, 364, 121–126. [Google Scholar] [CrossRef]
- Grimvall, G. Reconciling ab initio and semiempirical approaches to lattice stabilities. Berichte Der Bunsenges. Für Phys. Chem. 1998, 102, 1083–1087. [Google Scholar] [CrossRef]
- Burton, B.P.; Dupin, N.; Fries, S.G.; Grimvall, G.; Fernandez-Guillermet, A.; Miodownik, P.; Oates, W.A.; Vinograd, V. Using Abinitio calculations in the Calphad enviroment. Z. Für Metallkunde. 2001, 92, 514–525. [Google Scholar]
- Lu, X.; Selleby, M.; Sundman, B. Theoretical modeling of molar volume and thermal expansion. Acta Mater. 2005, 53, 2259–2272. [Google Scholar] [CrossRef]
- Zhang, B.; Li, X.; Li, D. Assessment of thermal expansion coefficient for pure metals. Calphad 2013, 43, 7–17. [Google Scholar] [CrossRef]
- Kaptay, G. Approximated equations for molar volumes of pure solid fcc metals and their liquids from zero Kelvin to above their melting points at standard pressure. J. Mater. Sci. 2015, 50, 678–687. [Google Scholar] [CrossRef]
- Vegard, Y.L. Yon Lo Vegard. Z. Für Phys. 1921, 5, 17–26. [Google Scholar] [CrossRef]
- Hafner, J. A note on Vegard’s and Zen’s laws. J. Phys. F Met. Phys. 1985, 15, 43–48. [Google Scholar] [CrossRef]
- Materials Project. Available online: https://www.materialsproject.org (accessed on 17 June 2020).
- Pearson, W.B. A Handbook of Lattice Spacings and Structures of Metals and Alloys, International Series of Monographs on Metal Physics and Physical Metallurgy Volume 4; Pergamon Press: Oxford, UK, 1958. [Google Scholar]
- Ellner, M.; Kattner, U.; Predel, B. Konstitutionelle und strukturelle untersuchungen im aluminiumreichen teil der systeme Ni-Al und Pt-Al. J. Less-Common Met. 1982, 87, 305–325. [Google Scholar] [CrossRef]
- Taylor, A.; Doyle, N.J. Further Studies on the Ni-Al System. I. The beta-NiAI and delta Ni2Al3 phase fields. J. Appl. Crystallogr. 1972, 5, 201. [Google Scholar] [CrossRef]
- Shockley, W. Theory of order for the copper gold alloy system. J. Chem. Phys. 1938, 6, 130–144. [Google Scholar] [CrossRef]
- Finel, A.; Ducastelle, F. On the phase diagram of the fcc ising model with antiferromagnetic first-neighbour interactions. Europhys. Lett. 1986, 1, 135–140. [Google Scholar] [CrossRef]
- Tétot, R.; Finel, A.; Ducastelle, F. Superdegenerate point in FCC phase diagram: CVM and Monte Carlo investigations. J. Stat. Phys. 1990, 61, 121–141. [Google Scholar] [CrossRef]
- Steiner, M.A.; Comes, R.B.; Floro, J.A.; Soffa, W.A.; Fitz-Gerald, J.M. L1′ ordering: Evidence of L10-L12 hybridization in strained Fe38.5Pd61.5 epitaxial films. Acta Mater. 2015, 85, 261–269. [Google Scholar] [CrossRef]
- Kusoffsky, A.; Sundman, B. Irregular Composition-Dependence of the Configurational Heat Capacity in the Modelling of Ordered Alloys. J. Phys. Chem. Solids 1998, 59, 1549–1553. [Google Scholar] [CrossRef]
- Geng, H.Y.; Sluiter, M.H.F.; Chen, N.X. Order-disorder effects on the equation of state for fcc Ni-Al alloys. Phys. Rev. B 2005, 72, 014204. [Google Scholar] [CrossRef]
- Bradley, A.J.; Taylor, A. An X-Ray Analysis of the Nickel-Aluminium System. Proc. R. Soc. Lond. A 1937, 159, 56–72. [Google Scholar]
- Van Der Ven, A. (Materials Department, University of California, Santa Barbara, CA, USA). Personal communication, 2019.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Entropy Model | TDB Available | Approach |
|---|---|---|---|
| This work | BWG | yes | DFT-CEF + no excess parameters reference state at 0 K |
| Al–Ni [9] | CE/MC | no | DFT 0 K |
| ATAT [13] | CE/CVM/MD | yes | DFT + finite temperature |
| Co-Ta [14] | BWG + excess | yes | DFT-CEF + excess parameters reference state at 298.15 K (SGTE) |
| Aluminium | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | A1 | A2 | A3 | A15 | A12() | D8() | C36 | C14 | C15 | D5 | D0 | |
| H (kJ/mol) | ||||||||||||
| This work | 0.0 | 9.3 | 3.2 | 7.7 | 5.1 | 6.3 | 10.0 | 15.0 | 14.3 | 15.6 | 17.1 | 1.4 |
| PAW-PW91 [24] | 0.0 | 9.7 | - | 7.5 | 4.9 | 6.5 | 9.7 | - | 13.9 | 15.1 | - | - |
| PAW-PW91 [25] | 0.0 | 9.21 | 2.85 | - | - | - | - | - | - | - | - | - |
| SGTE [26] | 0.0 | 10.1 | 5.5 | - | - | - | - | - | - | - | - | - |
| V (Å/at) | ||||||||||||
| This work | 16.48 | 16.91 | 16.64 | 16.93 | 17.13 | 16.91 | 17.31 | 17.45 | 17.45 | 17.43 | 18.78 | 16.59 |
| PAW-PBE [27] | 16.71 | - | - | - | - | - | - | - | - | - | - | - |
| FPLMTO-GGA [28] | 16.63 | - | - | - | - | - | - | - | - | - | - | - |
| LMTO-LDA [29] | 15.93 | 16.17 | - | - | - | - | - | - | - | - | - | - |
| Calphad [30] | - | 17.13 | 16.85 | - | - | - | - | - | - | - | - | - |
| Calphad [31] | 16.30 | 15.50 | 15.50 | - | - | - | - | - | - | - | - | - |
| B (GPa) | ||||||||||||
| This work | 77.6 | 69.8 | 73.3 | 64.7 | 69.4 | 74.0 | 70.1 | 65.7 | 66.3 | 66.5 | 67.4 | 76.0 |
| PAW-PBE [27] | 72.5 | - | - | - | - | - | - | - | - | - | - | - |
| FPLMTO-GGA [28] | 74.4 | - | - | - | - | - | - | - | - | - | - | - |
| LMTO-LDA [29] | 87.0 | 88.0 | - | - | - | - | - | - | - | - | - | - |
| Exp. [32] | 72.7 | - | - | - | - | - | - | - | - | - | - | - |
| Nickel | ||||||||||||
| Ref. | A1 | A2 | A3 | A15 | A12() | D8() | C36 | C14 | C15 | D5 | D0 | |
| H (kJ/mol) | ||||||||||||
| This work | 0.0 | 5.3 | 2.7 | 11.2 | 8.3 | 8.4 | 14.1 | 17.5 | 17.0 | 18.2 | 30.5 | 5.8 |
| PAW-GGA [24] | 0.0 | 8.8 | - | 12.7 | 9.1 | 16.5 | 16.0 | - | 19.0 | 21.3 | - | - |
| PAW-GGA [25] | 0.0 | 9.15 | 2.13 | - | - | - | - | - | - | - | - | - |
| SGTE [26] | 0.0 | 8.7 | 2.9 | - | - | - | - | - | - | - | - | - |
| V (Å/at) | ||||||||||||
| This work | 10.88 | 10.94 | 10.92 | 11.11 | 11.07 | 11.08 | 11.30 | 11.40 | 11.39 | 11.43 | 12.20 | 11.08 |
| PAW-PBE [27] | 10.90 | - | - | - | - | - | - | - | - | - | - | - |
| PAW-PBE [33] | 10.94 | - | - | - | - | - | - | - | - | - | - | - |
| LMTO-LDA [29] | 10.53 | 10.62 | - | - | - | - | - | - | - | - | - | - |
| B (GPa) | ||||||||||||
| This work | 199.3 | 193.7 | 189.7 | 185.3 | 202.1 | 189.5 | 177.2 | 188.5 | 198.5 | 183.5 | 165 | 145.2 |
| PAW-PBE [27] | 190.5 | - | - | - | - | - | - | - | - | - | - | - |
| PAW-PBE [33] | 193.0 | - | - | - | - | - | - | - | - | - | - | - |
| LMTO-LDA [29] | 248.0 | 224.0 | - | - | - | - | - | - | - | - | - | - |
| x(Ni) | Strukturbericht | Formation Energy [kJ/mol at] | Pearson Symbol | Space Group | Wyckoff Positions |
|---|---|---|---|---|---|
| fcc related | |||||
| 0.0 | A1-BB | 0.0 | cF4 (Cu) | Fmm (225) | 4a, 4b, 8c |
| 0.111 | Al8Ni-AB8 | −10.3 | tI18 (NbNi) | I4/mmm | 2a, 8h, 8i |
| 0.125 | D1D7-AB7 | −10.3 | cF32(CaGe) | Fmm (225) | 4a, 4b, 24d |
| 0.25 | D0-AB3 | −22.3 | tI8 (AlTi) | I4/mmm (139) | 2a, 2b, 4d |
| 0.25 | L1-AB3 | −22.3 | cP4 (CuAu) | Pmm (227) | 1a, 3c |
| 0.25 | D0-AB3 | −22.8 | tI16 (AlZr) | I4/mmm (139) | 4c, 4d, 4e |
| 0.25 | L6-AB3 | −22.3 | tP4 (CuTi) | P4/mmm (123) | 1a 1c 2e 2e |
| 0.333 | 12-AB2 | −38.5 | tI6 | I4/mmm (139) | - |
| 0.5 | CH40-AB | −58.2 | tI8(NbP) | I4/amd (141) | 2a, 2b |
| 0.5 | D4-AB | −31.0 | - | Fdm | - |
| 0.5 | L1-AB | −52.4 | tP2 (CuAu) | P4/mmm (123) | 1a, 1d |
| 0.5 | L1-AB | −43.0 | hR32 (CuPt) | Rm (166) | 1a, 1b |
| 0.5 | Z2-AB | −39.3 | tP8 | P4/nmm (129) | |
| 0.667 | 12-A2B | −45.6 | tI6 | I4/mmm (139) | |
| 0.75 | D0-A3B | −43.1 | tI8 (AlTi) | I4/mmm (139) | 2a, 2b, 4d |
| 0.75 | L1-A3B | −45.4 | cP4 (CuAu) | Pmm (227) | 1a, 3c |
| 0.75 | L6-A3B | −45.0 | tP4 (CuTi) | P4/mmm (123) | 1a 1c 2e 2e |
| 0.875 | D1D7-A7B | −23.2 | cF32(CaGe) | Fmm (225) | 4a, 4b, 24d |
| 0.889 | Al8Ni-A8B | −21.1 | tI18 (NbNi) | I4/mmm | 2a, 8h, 8i |
| 1.0 | A1-AA | 0.0 | cF4 (Cu) | Fmm (225) | 4a, 4b, 8c |
| bcc related | |||||
| 0.0 | A2-BB | +9.3 | cI2 (W) | Imm (229) | 2a |
| 0.25 | D0-AB3 | −9.6 | cF16 (AlFe) | Fmm (225) | 4a, 4b, 8c |
| 0.333 | C11-AB2 | −39.6 | tl6 | I4/mmm (139) | 2a, 4e |
| 0.375 | PdTi-A3B5 | −47.0 | - | - | - |
| 0.4 | AlOs-A2B3 | −51.5 | - | - | - |
| 0.428571 | B11-A3B4 | −45.1 | tP4 (CuTi) | P4/nmm (129) | 2c |
| 0.444 | VZn-A4B5 | −31.5 | tI18 (VZn5) | I4/mmm (139) | 2a, 8h, 8i |
| 0.5 | B2-AB | −66.3 | cP2 (CsCl) | Pmm (221) | 1a, 1b |
| 0.5 | B32-AB | −35.6 | cF16 (NaTl) | Fdm (227) | 8a, 8b |
| 0.555556 | VZn-A5B4 | −43.7 | tI18 (VZn5) | I4/mmm (139) | 2a, 8h, 8i |
| 0.571429 | B11-A4B3 | −43.0 | tP4 (CuTi) | P4/nmm (129) | 2c |
| 0.6 | AlOs-A3B2 | −55.7 | - | - | - |
| 0.625 | PdTi-A5B3 | −52.3 | - | - | - |
| 0.667 | C11-A2B | −46.8 | tl6 | I4/mmm (139) | 2a, 4e |
| 0.75 | D0-A3B | −40.9 | cF16 (AlFe) | Fmm (225) | 4a, 4b, 8c |
| 1.0 | A2-AA | +5.3 | cI2 (W) | Imm (229) | 2a |
| hcp related | |||||
| 0.0 | A3-BB | +3.2 | hP2 (Mg) | P6/mmc (194) | 2c |
| 0.25 | D0-AB3 | −21.2 | hP8 (NiSn) | P6/mmc (194) | 2c, 6h |
| 0.333 | B8-AB2 | −33.3 | hP6 (InNi) | P6/mmc (194) | 2a, 2c, 2d |
| 0.333 | B-AB2 | −41.0 | hP9 (AgZn) | P (147) | 1a, 2d, 6g |
| 0.5 | hcp-AB | −48.2 | hP2 | Pm2 (187) | 1a, 1f |
| 0.5 | B-AB | −44.5 | hP2 (WC) | Pm2 (187) | 1a, 1f |
| 0.5 | B-BA | −44.5 | hP2 (WC) | Pm2 (187) | 1a, 1f |
| 0.5 | B-AB | −19.0 | hP8 (AsTi) | P6/mmc (194) | 2a, 2d, 4f |
| 0.5 | B-BA | −49.8 | hP8 (AsTi) | P6/mmc (194) | 2a, 2d, 4f |
| 0.5 | B8-BA | −10.3 | hP4 (NiAs) | P6/mmc (194) | 2a, 2c |
| 0.5 | B35-BA | −2.2 | hP6 (CoSn) | P6/mmm (191) | 1a, 2d, 3f |
| 0.667 | B-A2B | −43.8 | hP9 (AgZn) | P (147) | 1a, 2d, 6g |
| 0.667 | B8-A2B | −50.0 | hP6 (InNi) | P6/mmc (194) | 2a, 2c, 2d |
| 0.75 | D0-A3B | −44.6 | hP8 (NiSn) | P6/mmc (194) | 2c, 6h |
| 1.0 | A3-AA | +2.7 | hP2 (Mg) | P6/mmc (194) | 2c |
| Phase | Pearson Symbol | Space Group | Wyckoff Positions | Configurations |
|---|---|---|---|---|
| D5 (AlNi) | hP5 (AlNi) | Pm1 (164) | 1a 2d 2d | 8 |
| D0 (AlNi) | oP16 (FeC) | Pmna (62) | 4c 4c 8d | 8 |
| TCP | ||||
| hR39 (FeW) | Rm (166) | 3a, 18h, 6c, 6c, 6c | 32 | |
| D8() | tP30 (FeCr) | P4/mnm (136) | 2a, 4f, 8i, 8i, 8j | 32 |
| A12() | cI58 ( Mg) | I3m (217) | 2a, 8c, 24g, 24g | 16 |
| A15 | cP8 (CrSi) | Pmm (223) | 6c, 2a | 4 |
| Laves | ||||
| C14 | hP12 (MgZn) | P6/mmc (194) | 2a, 4f, 6h | 8 |
| C15 | cF24 (MgCu) | Fdm (227) | 16d, 8a | 4 |
| C36 | hP24 (MgNi) | P6/mmc (194) | 4e, 4f, 4f, 6g, 6h | 32 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tumminello, S.; Palumbo, M.; Koßmann, J.; Hammerschmidt, T.; Alonso, P.R.; Sommadossi, S.; Fries, S.G. DFT-CEF Approach for the Thermodynamic Properties and Volume of Stable and Metastable Al–Ni Compounds. Metals 2020, 10, 1142. https://doi.org/10.3390/met10091142
Tumminello S, Palumbo M, Koßmann J, Hammerschmidt T, Alonso PR, Sommadossi S, Fries SG. DFT-CEF Approach for the Thermodynamic Properties and Volume of Stable and Metastable Al–Ni Compounds. Metals. 2020; 10(9):1142. https://doi.org/10.3390/met10091142
Chicago/Turabian StyleTumminello, Silvana, Mauro Palumbo, Jörg Koßmann, Thomas Hammerschmidt, Paula R. Alonso, Silvana Sommadossi, and Suzana G. Fries. 2020. "DFT-CEF Approach for the Thermodynamic Properties and Volume of Stable and Metastable Al–Ni Compounds" Metals 10, no. 9: 1142. https://doi.org/10.3390/met10091142
APA StyleTumminello, S., Palumbo, M., Koßmann, J., Hammerschmidt, T., Alonso, P. R., Sommadossi, S., & Fries, S. G. (2020). DFT-CEF Approach for the Thermodynamic Properties and Volume of Stable and Metastable Al–Ni Compounds. Metals, 10(9), 1142. https://doi.org/10.3390/met10091142