Abstract
In order to prepare an Fe(II) spin crossover (SCO) complex that could be consequently modified to a bimetallic coordination compound that possesses another magnetic property of interest, a specially designed ligand L-NH2 (1-(4-aminobenzyl)-4,11-bis(pyridine-2-ylmethyl)- 1,4,8,11-tetraazacyclotetradecane) was prepared. This ligand consists of a macrocyclic cyclam part containing two 2-pyridylmethyl pendant arms (expecting SCO upon Fe(II) complexation) and one p-aminobenzyl pendant arm with an NH2 group. The presence of this group enables the consequent transformation to various functional groups for the selective complexation of other transition metals or lanthanides (providing the second property of interest). Furthermore, the performed theoretical calculations (TPSSh/def2-TZVP) predicted SCO behavior for the Fe(II) complex of L-NH2. Thus, Fe(II) complexes [Fe(L-NH2)](ClO4)2 (1) and [Fe(L-NH2)]Cl2·6H2O (2) were synthesized and thoroughly characterized. Based on the crystal structure of an isostructural analogous Ni(II) complex [Ni(L-NH2)]Cl2·6H2O (3), the coordination number six was confirmed with an octahedral coordination sphere and a cis-arrangement of the pyridine pendant arms. The measured magnetic data confirmed the high-spin behavior of both compounds with large magnetic anisotropy (D = 17.8 for 1 and 20.9 cm−1 for 2 complemented in both cases also with large rhombicity), though unfortunately without any indication of the SCO behavior with decreasing temperature. The lack of SCO can be ascribed to the crystal packing and/or the non-covalent intermolecular interactions stabilizing the high-spin state in the solid state.
1. Introduction
The spin crossover (SCO) phenomenon is a modern, fascinating, and rapidly developing topic of interest in the field of molecular magnetism. SCO compounds show a reversible transition between the high-spin (HS) and low-spin (LS) magnetic states that is induced by different chemical (e.g., ligand and solvent effects) or, more often, physical external stimuli (e.g., temperature, light irradiation (LIESST = light-induced electron spin-state trapping) [1,2], pressure, or magnetic field) [3]. If SCO is induced by temperature, the material is commonly characterized by its transition temperature (T1/2) and by the abruptness of the transition between the HS and LS states that depends on the cooperative effect. Many remarkable applications can be found for SCO compounds, e.g., various electronic device miniaturization applications including high capacity memories, low-energy demanding displays, or molecular switches [4].
Recently, this field has been expanded to the investigation of multifunctional magnetic materials combining SCO with one or more properties of interest, e.g., electric conductivity, non-linear optics, luminescence, magnetic exchange coupling, or long-range magnetic ordering [4,5]. Additionally, the coupling of SCO with single-molecular magnetism appears to be very interesting. So-called single-molecule magnets (SMMs) [6,7] are chemical compounds whose relaxation of magnetization is slow and has a molecular origin (there is no long-range ordering as typically observed in bulk magnets); therefore, they can be classified as “nanomagnets”. Moreover, these compounds have extensive application potential in, e.g., high-density storage media, quantum computing, or spintronics [8]. Surprisingly, there still exists very little information about compounds that combine both SCO and SMM, especially in one discrete molecule, and some completely new fundamental questions, e.g., what is the influence of the spin transition on the SMM properties, and the relaxation mechanism (s), remain open. Previously, several strategies for the development of such multifunctional compounds combining SCO with SMM properties have already been employed. One methodology was based on the coupling of two units, each having the appropriate property, by using a simple non-specific linker in a 1D chain [9]. Another strategy used one mononuclear Fe(II) or Fe(III) complex exhibiting both SCO and SMM properties [10,11,12,13] or the co-crystallization/metal dilution of Fe(II) SCO centers with Co(II) field-induced SMM centers [14].
Our long-term aim is to prepare one discrete molecule/polynuclear complex that contains a rationally designed ligand with at least two specific coordination sites that are capable of the complexation of Fe(II) (providing SCO) and either lanthanides or other transition metals (providing, e.g., SMM properties). This approach is rather demanding because, more-or-less, advanced organic syntheses are usually required. Moreover, the presence/absence of SCO and its abruptness are very sensitive to any structural modification of the parent ligand in the Fe(II) SCO complex, because such modification can very often influence the ligand field, non-covalent contacts, and the final crystal packing. Such structural changes have been documented on several ligand families derived from, e.g., diimines (bipyridine, phenanthroline, and terpyridine) [15], 2,6-bis(pyrazolyl)pyridines (bpp) [16,17,18], or Schiff bases [19,20,21].
The initial idea for the preparation of the rationally designed ligand in this work was based on the fact that cyclam substituted with two pendant arms that contain the pyridine moiety (Py2-C, Figure 1) rather independently provides SCO in its Fe(II) complexes [Fe(Py2-C)](BF4)2·H2O (T1/2 ~ 150 K) [22], [Fe(Py2-C)](C(CN)3)2·2H2O (T1/2↓/T1/2↑ = 136/145 K), and [Fe(Py2-C)][Ni(CN)4]·H2O (T1/2↓↑ = 85 K) [23] on the counter anion. The same holds for Fe(II) complexes of the cross-bridged analogue Py2-CB [24] (Figure 1) containing various counter anions ([FeCl4]2−, Cl−, BF4−, BPh4−), in which SCO is preserved, although the original NH groups were replaced by the ethylene bridge.
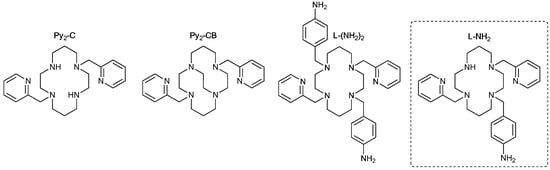
Figure 1.
Structural formulas of the herein investigated ligand L-NH2, together with other ligands previously used for the preparation of the Fe(II) complexes discussed in the text.
Therefore, in our previous work, ligand L-(NH2)2 (Figure 1), in which two macrocyclic nitrogen atoms of the Py2-C part were modified with the p-aminobenzyl group, was prepared and characterized [25]. Such structural modification was chosen because (i) the disubstituted product was more simply available from the synthetic point of view, and (ii) the NH2 group in the p-aminobenzyl pendant arms could be consequently easily converted into different functional groups for the selective coordination of, e.g., lanthanides (the introduction of SMM properties). However, before the realization of such a transformation of the NH2 group, it was necessary to investigate the magnetic properties of the Fe(II) complex of L-(NH2)2. Unfortunately, for the prepared Fe(II) complex [Fe(L-(NH2)2)]Cl2·2.5H2O, SCO was not observed. According to the performed theoretical calculations, the reason behind the absence of SCO was very likely an insufficiently strong ligand field that was reduced by the electron-withdrawing effect of the two benzyl groups. In the next step, theoretical density functional theory (DFT) calculations were performed for the Fe(II) complexes with a monosubstituted L-NH2 ligand containing only one p-aminobenzyl pendant arm (Figure 1), which would partially eliminate weakening of the ligand field. Based on these calculations, which suggested the possibility of the SCO behavior of such complexes, the monosubstituted L-NH2 ligand was synthesized along with its Fe(II) complexes with different counter anions. Their structural and magnetic properties were investigated and are discussed in detail in this work.
2. Materials and Methods
The ligand Py2-C [26] was synthesized according to the literature procedure. All used solvents (VWR International) and chemicals were purchased from commercial sources (Across Organics or Sigma-Aldrich), and they were used without any further purification.
An elemental analysis (C, H, and N) was carried out on a Flash 2000 CHNO-S analyzer (Thermo Scientific, Waltham, MA, USA). 1H and 13C NMR spectra were recorded on a 400-MR NMR spectrometer (Varian, Palo Alto, CA, USA) at 25 °C: 1H 399.95 MHz, chloroform-d (CDCl3, tetramethylsilane or residual solvent peak) δ = 0.00 or 7.27 ppm, respectively, 13C 100.60 MHz, (CDCl3, residual solvent peak) δ = 77.0 ppm. The multiplicity of the signals is indicated as follows: s—singlet; d—doublet; quin—quintet; and m—multiplet. A deuterated solvent CDCl3 from Sigma-Aldrich was used as received. The scheme of atom numbering that was used for NMR data interpretation is displayed in Figure 2. The hydrogen and carbon atoms were assigned according to the two-dimensional correlation spectra 1H–13C gs-HMQC (heteronuclear multiple-quantum correlation) and 1H–13C gs-HMBC (heteronuclear multiple-bond correlation, see Figures S1–S4 in Supplementary Materials). The mass spectra were recorded on an LCQ Fleet mass spectrometer (Thermo Scientific, Waltham, MA, USA) that used electrospray ionization (ESI) and was equipped with a three-dimensional ion-trap detector in both positive and negative modes. A Jasco FT/IR-4700 spectrometer (Jasco, Easton, MD, USA) was used for the collection of the infrared (IR) spectra of the studied ligand and complexes in the range of 400−4000 cm−1 by using the attenuated total reflection (ATR) technique on a diamond plate. The temperature dependence of the magnetization at B = 0.1 T from 1.9 to 300 K and field dependence data at T = 2.0 and eventually at T = 4.6 K (direct current (DC) magnetic data) were measured by using MPMS-SQUID-XL7 (Magnetic Properties Measurement System—Superconducting Quantum Interference Device, Quantum Design Inc., San Diego, CA, USA). The experimental data were corrected for diamagnetism and the signal of the sample holder. The X-ray powder diffraction patterns were recorded on a MiniFlex600 (Rigaku) diffractometer by using Cu Kα radiation (λ = 1.5418 Å). The molecular structure of the studied complex that is depicted in Figure 3 and Figure S6 was drawn by using the Mercury software 4.3.0 [27].
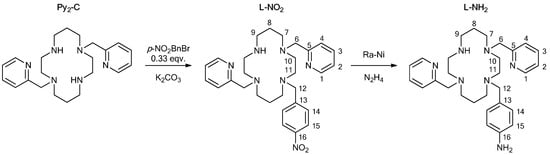
Figure 2.
Synthetic scheme of the preparation of the ligand L-NH2 together with atom numbering of both L-NO2 and L-NH2 used in 1H/13C NMR data interpretation.
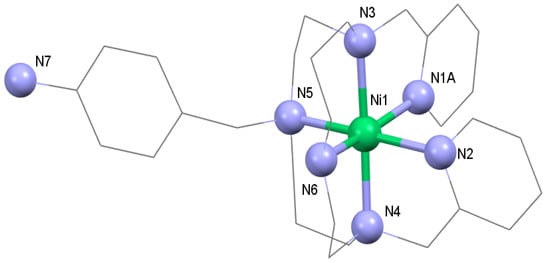
Figure 3.
Molecular structure of the [Ni(L-NH2)]2+ cation that was found in the crystal structure of complex 3. The disordered parts of the molecule and hydrogen atoms are omitted for clarity.
2.1. Synthesis
2.1.1. Ligand
1-(4-nitrobenzyl)-4,11-bis(pyridine-2-ylmethyl)-1,4,8,11-tetraazacyclotetradecane (L-NO2)
Macrocycle Py2-C (4.000 g, 10.46 mmol, 3.0 eqv.) and K2CO3 (2.500 g, 18.09 mmol, 5.2 eqv.) were suspended in 80 mL of CH3CN and heated to reflux. p-nitrobenzyl bromide (754 mg, 3.49 mmol) dissolved in 20 mL of CH3CN was added to this refluxing mixture via a syringe pump (1 mL/min). The obtained mixture was refluxed for further 20 min and then cooled down to room temperature. The reaction mixture was controlled by the measurement of a mass spectrum that contained the signals of the parent macrocycle Py2-C and only the monosubstituted product. The suspension was filtered on a glass frit, and the obtained filtrate was evaporated in vacuo. The crude product was purified with gradient column chromatography (SiO2, CHCl3/CH3OH/25% aq. NH3 10/2/0.2 (Rf = 0.4) → 10/10/2). Collected fractions containing the product were put together, and the solvents were evaporated under reduced pressure. The pure product was isolated in the form of light-yellow oil (1.53 g, yield 84.7%).
MS m/z (+): 518.39 ([L + H]+, calcd. 518.32).
1H NMR (CDCl3): δ 1.69 (2H, quin, 3JHH = 6.0 Hz, H-8), 1.80 (2H, quin, 3JHH = 6.0 Hz, H-8), 2.47–2.80 (16H, m, H-7,9,10,11), 3.46 (2H, s, H-12), 3.69 (2H, s, H-6), 3.72 (2H, s, H-6), 7.10 (2H, m, H-2), 7.38 + 7.42 (2H, m, H-4), 7.44 (2H, m, AA’BB’, H-14), 7.51–7.60 (2H, m, H-3), 8.08 (2H, m, AA´BB´, H-15), 8.47 (2H, m, H-1).
13C{1H} NMR (CDCl3): δ 25.27 + 25.66 (2C, C-8), 46.61 + 47.45 + 49.63 + 51.38 + 51.59 + 51.90 + 52.57 + 53.54 (8C, C-7,9,10,11), 57.81 (1C, C-12), 59.84 (1C, C-6), 60.76 (1C, C-6), 121.79 (2C, C-2), 122.95 + 123.27 (2C, C-4), 123.20 (2C, C-15), 129.27 (4C, C-14), 136.02 + 136.17 (2C, C-3), 146.76 (1C, C-16), 148.04 (1C, C-13), 148.83 (2C, C-1), 159.15 + 159.35 (2C, C-5).
1-(4-aminobenzyl)-4,11-bis(pyridine-2-ylmethyl)-1,4,8,11-tetraazacyclotetradecane (L-NH2)
The synthesis of L-NH2 was performed according to the literature procedure [25]. L-NO2 (1.53 g, 2.96 mmol) was reduced by hydrazine monohydrate (3.00 g, 59 mmol) and Raney-Ni (Ra-Ni) in refluxing CH3OH. The reaction progress was monitored by measurements of mass spectra. The product was isolated as light-yellow oil (1.35 g, yield 93.8%).
MS m/z (+): 488.35 ([L + H]+, calcd. 488.35).
1H NMR (CDCl3): δ 1.73–1.81 (4H, m, H-8), 2.51–2.80 (16H, m, H-7,9,10,11), 3.33 (2H, s, H-12), 3.60 (2H, s, NH2), 3.73 (2H, s, H-6), 3.76 (2H, s, H-6), 6.60 (2H, m, AA´BB´, H-15), 7.04 (2H, m, AA´BB´, H-14), 7.13 (2H, m, H-2), 7.41 (1H, d, 3JHH = 7.8 Hz, H-4), 7.51 (1H, d, 3JHH = 7.8 Hz, H-4), 7.61 (2H, m, H-3), 8.52 (2H, m, H-1).
13C{1H} NMR (CDCl3): δ 25.13 (2C, C-8), 46.68 + 47.81 + 49.66 + 51.25 + 51.68 + 52.61 (8C, C-7,9,10,11), 57.68 (1C, C-12), 60.09 (1C, C-6), 60.78 (1C, C-6), 114.81 (2C, C-15), 121.82 (2C, C-2), 123.09+123.47 (2C, C-4), 129.07 (1C, C-13), 130.19 (2C, C-14), 136.19+136.32 (2C, C-3), 145.05 (1C, C-16), 148.87 (2C, C-1), 159.57 (2C, C-5).
2.1.2. Complexes 1–3
[Fe(L-NH2)](ClO4)2 (1)
Compound L-NH2 (100 mg, 0.205 mmol) was dissolved in 2.0 mL of hot methanol. Fe(ClO4)2·6H2O (74 mg, 0.205 mmol) was dissolved in deionized water (2 mL) together with a small amount of ascorbic acid, thus preventing air-oxidation. Both solutions were mixed and heated to reflux for 5 min. During that time, the dark green solution turned yellow, and a light yellow precipitate started to form. The mixture was cooled down to 5 °C in a fridge and filtered on a glass frit, and the solid was washed with acetone (2 mL). The product was obtained in the form of a light-yellow microcrystalline powder (77 mg, yield 50.6%).
MS m/z (+): 542.27 ([Fe(L-NH2–H+)]+, calcd. 542.27), 642.22 ([Fe(L-NH2) + (ClO4)–]–, calcd. 642.23).
MS m/z (–): 840.31 ([Fe(L-NH2) + 3(ClO4)−]−, calcd. 840.12).
Anal. Calcd. (%) for [Fe(L-NH2)](ClO4)2 (C29H41Cl2FeN7O8, Mr = 742.43): C, 46.91; H, 5.57; N, 13.21. Found: C, 46.54; H, 5.49; N, 12.70.
Caution!Though we experienced no difficulties, perchlorate salts of metal complexes with organic ligands are potentially explosive and should be handled with great care even in small quantities.
[Fe(L-NH2)]Cl2·6H2O (2)
Anhydrous FeCl2 (52 mg, 0.411 mmol) was dissolved in methanol (2 mL) together with a small amount of ascorbic acid (dissolved in several drops of deionized water) added against air-oxidation. Compound L-NH2 (200 mg, 0.411 mmol) was dissolved in methanol (4 mL). Both solutions were mixed and heated to reflux for 5 min. During that time, the dark green solution turned green-yellow. Then, the solution was cooled down to room temperature. The brown solid, which formed during the procedure, was filtered via a syringe filter. Diethyl ether (60 mL) was slowly (over approximately 20 min) added to the green-yellow filtrate, which induced the precipitation of a brown-green-yellow solid. The obtained solid was decanted with diethyl ether (20 mL) and dried by heating to 60 °C for 5 min. The product was obtained in the form of a yellow-green powder (180 mg, yield 60.6%).
MS m/z (+): 542.28 ([Fe(L-NH2–H+)]+, calcd. 542.27), 578.26 ([Fe(L-NH2) + Cl–]+, calcd. 578.25).
Anal. Calcd. (%) for [Fe(L-NH2)]Cl2·6H2O (C29H53Cl2FeN7O6, Mr = 722.53): C, 48.21; H, 7.39; N, 13.57. Found: C, 48.57; H, 7.27; N, 13.78.
[Ni(L-NH2)]Cl2·6H2O (3)
Ligand L-NH2 (70 mg, 0.144 mmol) together with NiCl2·6H2O (34 mg, 0.143 mmol) was dissolved in 4 mL of methanol and heated to reflux for 5 min. The obtained pink solution was filtered via a syringe filter and crystallized by diffusion of diethyl ether vapors at 5 °C. The product was isolated in the form of violet crystals (25 mg, yield 24.1%).
MS m/z (+): 544.40 ([Ni(L-NH2–H+)]+, calcd. 544.27), 580.25 ([Ni(L-NH2) + Cl–)]–, calcd. 580.25). MS m/z (–): 1269.64 ([(Ni(L-NH2))2 + 5Cl–)]–, calcd. 1269.39).
Anal. Calcd. (%) for [Ni(L-NH2)]Cl2·6H2O (C29H53Cl2NiN7O6, Mr = 725.37): C, 48.02; H, 7.36; N, 13.52. Found: C, 48.47; H, 7.14; N, 13.38.
2.2. Theoretical Calculations
The theoretical calculations were performed by using the ORCA 4.1 software [28]. The hybrid meta-GGA functional TPSSh [29,30] together with a polarized triple-ζ quality basis set def2-TZVP [31] for all atoms were employed for the molecular geometry optimization procedure, in which tight optimization criteria were set in ORCA. All calculations exploited the resolution of identity approximation with the decontracted auxiliary def2/J Coulomb fitting basis set [32] and the chain-of-spheres (RIJCOSX = resolution of identity approximation for Coulomb integrals and COSX numerical integration) approximation to exact exchange [33,34]. The integration grids were increased by setting Grid5 and Gridx5, and the convergence criteria were set to tight SCF (self-consistent field) in all calculations.
3. Results and Discussion
3.1. Syntheses and General Characterizations
The studied ligand L-NH2 was prepared in the two steps that are depicted in Figure 2 in the same way as described in the literature [25]. In the first step, the derivative L-NO2 was prepared with a common SN2 substitution reaction of the parent Py2-C macrocycle with p-nitrobenzyl bromide. In order to obtain only the monosubstituted product, a three-fold molar excess of Py2-C had to be employed together with a consequent purification by column chromatography. The reaction mixture was checked by mass spectrometry. The signals of the parent macrocycle Py2-C and only the monosubstituted product were detected (Figure S5 in Supplementary Materials); nevertheless, a small amount of the disubstituted side-product was found in the first fractions during the chromatographic purification.
In the next step, the aromatic nitro group was reduced to an amino group by using the Ra-Ni catalyst and hydrazine hydrate as the source of hydrogen [25]. The reaction progress was controlled by mass spectrometry. The reduction was completed when the signal of the hydroxylamine intermediate vanished (two-to-three more additions of Ra-Ni had to be used in order to complete the reaction). In the mass spectrum of L-NH2 (Figure S5), the signal of the parent macrocycle Py2-C was also identified as the consequence of the fragmentation of the molecular ion corresponding to [L-NH2 + H+]+, because the cleavage ability of the p-aminobenzyl group during ESI was rather strong.
The Fe(II) complex 1 was synthesized through the direct mixing of a methanolic solution of L-NH2 with an aqueous solution of Fe(ClO4)2·6H2O and heating to reflux for 5 min. The product precipitated from the solution in the form of a light-yellow microcrystalline powder. Fe(II) complex 2 containing chloride counter anions was prepared by a similar procedure as for complex 1, but due to the higher solubility of complex 2, methanolic solutions of the ligand and FeCl2 were used, and the precipitation of the solid product had to be induced by addition of diethyl ether. Finally, complex 3, as an Ni(II) analogue of complex 2, was prepared as well. This complex was the only one that we were able to prepare in the form of single crystals suitable for X-ray diffraction analysis in spite of many attempts to obtain single crystals of the Fe(II) complexes by (i) the slow diffusion of diethyl ether vapors into their solutions at low temperature, (ii) the slow cooling of the complex solutions, and (iii) the slow evaporation or the complex solutions at room temperature. Unfortunately, the prepared crystals of complex 3 were only of poor quality, and the refined structural model was not acceptable for publication (R1 > 0.10, see Supplementary Materials, Figure S6 [27,35]). The structure exhibited whole-molecule disorder, which was only possible to partially establish for both pyridine pendant arms, while we failed in attempts to model the aliphatic parts of the ligand as disorder over two positions (Figure S6). Therefore, the corresponding atoms in the aliphatic parts exhibited very large anisotropic displacement parameters. Nevertheless, the best structural model that was obtained is shown in Figure 3, from which the molecular connectivity in [Ni(L-NH2)]2+ could be clearly seen. The Ni(II) central atom had a distorted octahedral geometry with the cis-arrangement of the pyridine pendant arms.
In order to confirm the isostructural character of complexes 2 and 3, a powder X-ray diffraction pattern was measured for these complexes, and this is shown in Figure 4. The similarity of both patterns clearly showed that both complexes were isomorphic and therefore probably isostructural, as well as the fact that the octahedral geometry with the cis-arrangement of the pyridine pendant arms was valid also for Fe(II) complex 2.
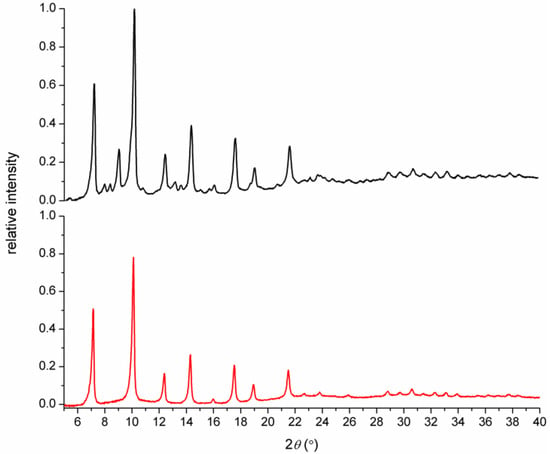
Figure 4.
Powder X-ray diffraction pattern measured for complexes 3 (black) and 2 (red).
Furthermore, the IR spectra of the ligand L-NH2 and the complexes 1–3 were measured as well (Figure S7). They showed the characteristic absorption bands corresponding to the NH valence vibrations (~3400 cm−1) and OH valence vibrations (~3350 and 3200 cm−1), thus confirming the presence of co-crystallized water molecules in the case of complexes 2 and 3, CH2 stretching vibrations (~2800 cm−1 for free ligand, which were shifted to ~2860 cm−1 upon its coordination), and stretching vibrations of the aromatic C=N and C=C bonds, as well as the CH2 bending and wagging vibrations of the pyridine ring (~1400, ~1500 and ~1600 cm−1). Strong signals corresponding to the presence of perchlorate anions (~1100 and ~600 cm−1) were found in the IR spectrum of complex 1.
3.2. Magnetic Measurements
The results of DC magnetic measurements for complexes 1 and 2 are displayed in Figure 5. The room temperature values of μeff/μB = 5.4 and 5.3 for complexes 1 and 2, respectively, were slightly higher in comparison to the theoretical spin-only value (μeff/μB = 4.90 for S = 2 and g = 2.0) because of a substantial contribution of the orbital angular momentum that gave rise to high magnetic anisotropy of the Fe(II) atom. Upon cooling, the μeff/μB values abruptly decreased below 50 K to a minimum of 2.9 or 3.1, respectively, at 1.9 K as a result of the zero-field splitting (ZFS) effects. The obtained data clearly showed that no SCO occurred and that both Fe(II) complexes remained in the HS state upon cooling down to 1.9 K. Such behavior is in contrast to the SCO Fe(II) complexes of Py2-C and Py2-CB, and it could be clarified on the basis of the following: (i) the electron-withdrawing effect of the benzyl group lowering the ligand field strength upon nitrogen donor atom substitution, (ii) changes in crystal packing and non-covalent intermolecular interactions, and (iii) the different crystallinity and morphology of the sample (lower cooperativity). On the other hand, the different counter anions have no observable effects on the presence/absence of SCO in the studied complexes 1 and 2. Complex 2 had six co-crystallized water molecules, while complex 1 had no solvent molecules; thus, the effect of solvation on the absence of SCO could play a rather minor role in this case.
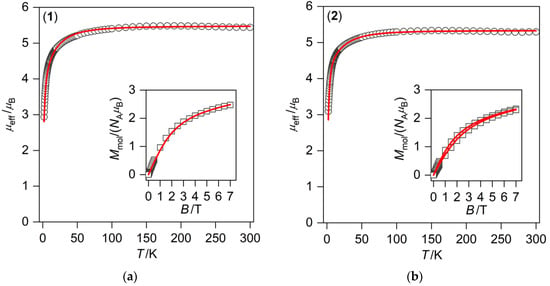
Figure 5.
Temperature dependence of the effective magnetic moment and the isothermal magnetizations measured at T = 2 and 4.6 K (inset) for (a) [Fe(L-NH2)](ClO4)2 (1); and (b) [Fe(L-NH2)]Cl2·6H2O (2). Empty symbols—experimental data; and full lines—calculated data with g = 2.24, D = 17.8 cm−1, E/D = 0.33, zj = –0.24 cm−1 (a) and g = 2.17, D = 20.9 cm−1, E/D = 0.31, zj = 0.24 cm−1 (b).
In the next step, the measured magnetic data were analyzed by using a spin Hamiltonian for mononuclear complexes
where the single-ion axial (D) and rhombic (E) ZFS parameters are incorporated [36] together with the Zeeman term defined for a-direction of a magnetic field as Ba = B(sin(θ)cos(φ), sin(θ)sin(φ), and cos(θ)) with the help of the polar coordinates. The solid state-induced intermolecular interactions are treated by the molecular-field correction parameter zj (where z is the number of the nearest neighbors and j is the interaction parameter that is taken in appropriate units) [36]. In the next step, the molar magnetization was calculated in the given direction of the magnetic field Ba:
where Za is the matrix element of the Zeeman term for the a-direction of the magnetic field and C represents the eigenvectors resulting from the diagonalization of the complete spin Hamiltonian matrix. The iterative procedure was applied due to the molecular-field correction (zj) [36]. Lastly, in order to properly simulate experimental powder magnetization data, the integral (orientational) average of molar magnetization was calculated as:
The best fit was found for the parameters g = 2.24, D = 17.8 cm−1, E/D = 0.33, zj = −0.24 cm−1 for complex 1 and g = 2.17, D = 20.9 cm−1, E/D = 0.31, zj = 0.24 cm−1 for complex 2 (Figure 5). The obtained ZFS-parameters were close to those found in compound [Fe(L-(NH2)2)]Cl2·2.5H2O (D = –17.7 cm−1, E/D = 0.31) [25] except for the sign of the D-parameter, which could be most likely related to the changes (symmetry and strength) in the ligand field, because, as we showed in another paper, the sign of the D-parameter is very sensitive to the modulation of the ligand field [37]. Such relatively high magnetic anisotropy is typical for HS FeII octahedral complexes [38].
SMM behavior, which is usually confirmed by the observation of an out-of-phase signal during an alternating current (AC) magnetic susceptibility measurement, is rather rare for d6 HS six-coordinate Fe(II) complexes [39]. In our case of compounds with positive D-values, the spin-reversal barrier (Ueff) for the slow relaxation of magnetization is not expected.
3.3. Theoretical Calculations
In our previous paper, DFT calculations were carried out in order to understand why the structural modification of Py2-C to L-(NH2)2 led to the transformation of the SCO behavior of [FeII(Py2-C)]2+ or [FeII(Py2-CB)]2+ compounds [22,23,24] into the HS state behavior of [Fe(L-(NH2)2)](BF4)2·2H2O [25]. Such behavior was justified by the presence of two benzyl groups with an electron-withdrawing effect that led to a weaker ligand field that was insufficient to trigger the SCO phenomenon. Therefore, herein, analogous DFT calculations were done with the hybrid meta-GGA functional TPSSh, which was recently benchmarked for SCO compounds [40]. The molecular geometries of [FeII(L-NH2)]2+ were optimized for both HS and LS states by using an ORCA computational package (TPSSh functional and def2-TZVP basis set for all atoms). The calculations were performed for both geometrical isomers, cis and trans, and the molecular geometries of the HS and LS isomers for cis-[FeII(L-NH2)]2+ are depicted in Figure 6. Indeed, the theoretical calculations confirmed our presumption that by removing one benzyl group, the ligand field became stronger, because the calculated energy separation of the HS and LS states, ∆ = εHS – εLS, now favored the LS state (Figure 7). Furthermore, the Δ parameters were in the same range as the SCO compounds [FeII(Py2-C)]2+ or [FeII(Py2-CB)]2+. It must also be noticed that in both the HS and LS states of [FeII(L-NH2)]2+, the cis-isomers were lower in energy (Figure 8), which was in accordance with the experimental structure of Ni(II) complex 3—Figure 3 and Figure S6. Therefore, all these theoretical calculations suggest that SCO behavior in [FeII(L-NH2)]2+ complex should be observed. On the contrary, the herein studied compounds 1 and 2 were found to be in the HS state in the whole temperature range of 2–300 K. A plausible explanation for this is that non-covalent interactions in solid state favor the HS configuration of these compounds despite the molecular predisposition of these complexes for SCO behavior.
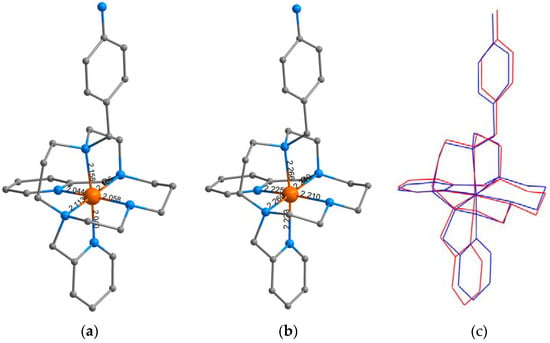
Figure 6.
(a) The density functional theory (DFT)-optimized low-spin (LS) state molecular geometry of cis-[Fe(L-NH2)]2+ by using TPSSh/def2-TZVP. (b) The DFT-optimized high-spin (HS) state molecular geometry of cis-[Fe(L-NH2)]2+ by using TPSSh/def2-TZVP. (c) The overlay of the DFT-optimized LS (blue) and HS (red) molecular geometries of cis-[Fe(L-NH2)]2+. The hydrogen atoms are omitted.
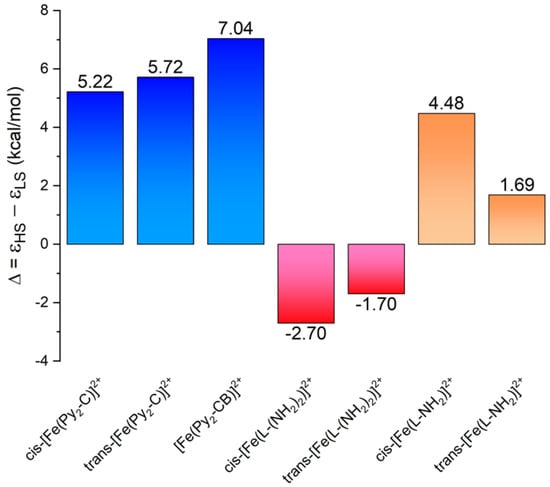
Figure 7.
The energy separation between the HS and LS states of cis/trans-[FeII(Py2-C)]2+, cis-[FeII(Py2-CB)]2+, and cis/trans-[Fe(L-(NH2)2)]2+ (data adopted from [25]) and cis/trans-[Fe(L-NH2)]2+ calculated by using TPSSh/def2-TZVP.
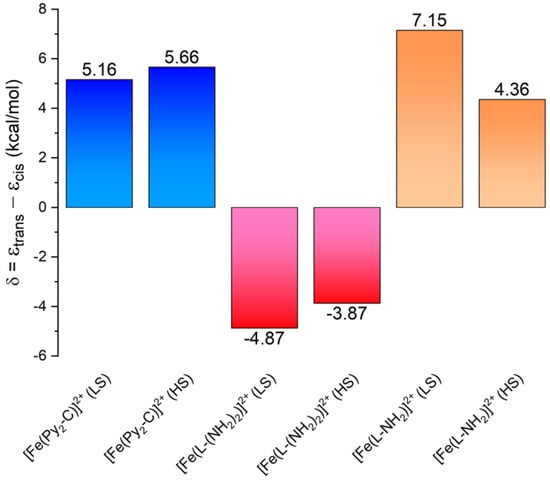
Figure 8.
The energy separation between the trans- and cis-isomers of the HS and LS forms of [FeII(Py2-C)]2+, [Fe(L-(NH2)2)]2+ (data adopted from [25]) and [Fe(L-NH2)]2+ calculated by using TPSSh/def2-TZVP.
Nevertheless, it should be stated here that the calculated results should be considered with great care because the applied functional may not be completely adequate for the full/correct reproduction of the energy level pattern of this particular molecular system and could be responsible for the observed discrepancy from the experimental data.
4. Conclusions
A rationally designed macrocyclic ligand L-NH2 containing an NH2 group, which allows for an easy structural transformation to other functional groups that are suitable for the selective complexation of 3d or 4f metal ions, was synthesized. Two Fe(II) complexes [Fe(L-NH2)](ClO4)2 and [Fe(L-NH2)]Cl2·6H2O were prepared, and the cis-arrangement of pyridine pendant arms in an octahedral coordination sphere was confirmed by the molecular structure of an isostructural Ni(II) analogue [Ni(L-NH2)]Cl2·6H2O. Based on the DC magnetic measurements, it was confirmed that both complexes remained in the HS state in the investigated temperature range and had a relatively high magnetic anisotropy, despite the fact that theoretical DFT calculations predicted the predisposition of this molecular system for SCO behavior analogously to compounds containing [FeII(Py2-C)]2+ [22,23] and [FeII(Py2-CB)]2+ [24].
We believed that the substitution of the parent macrocycle Py2-C, not with two (as in case of L-(NH2)2 [25]) but only with one p-aminobenzyl pendant arm (L-NH2), would result in increase of the ligand field and the presence of SCO behavior. Unfortunately, the prepared Fe(II) complexes remained in the HS state, and, thus, it can be concluded that other factors besides the ligand field strength play a significant role in this case, most likely the crystal packing and the intermolecular interactions and possibly the solvation as well. In conclusion, the utilization of p-aminobenzyl pendant arm(s) appeared to be unsuitable in this particular case of Py2-C modification. Nevertheless, there is still strong potential in the development of bifunctional ligand(s) to enable a combination of SCO with other property of interest in one discrete polynuclear complex. However, such strategy remains an immense chemistry challenge.
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-4701/10/3/366/s1, Figure S1: 1H–13C gs-HMQC NMR spectrum of L-NO2; Figure S2: 1H–13C gs-HMBC NMR spectrum of L-NO2; Figure S3: 1H–13C gs-HMQC NMR spectrum of L-NH2; Figure S4: 1H–13C gs-HMBC NMR spectrum of L-NH2; Figure S5: Mass spectra of the reaction mixtures during preparation of L-NO2 and L-NH2; Figure S6: Molecular structure of [Ni(L-NH2)]2+ cation found in the crystal structure of 3; Figure S7: Comparison of IR spectra measured for pure ligand L-NH2 as well as for its studied complex 1–3; X-ray diffraction analysis. Optimized xyz coordinates for cis/trans-[Fe(L-NH2)]2+ in HS and LS states.
Author Contributions
Conceptualization, B.D.; methodology, B.D. and R.H.; validation, B.D., R.H. and I.Š.; formal analysis, R.H.; investigation, B.D., P.A. and I.S.; data curation, P.A. and I.Š.; writing—original draft preparation, B.D. and R.H.; writing—review and editing, B.D., P.A., I.Š. and R.H.; visualization, B.D., P.A. and R.H.; supervision, B.D.; funding acquisition, I.Š. and R.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Grant Agency of the Czech Republic (GACR), grant number 17-08992S (Bohuslav Drahoš, Peter Antal, Radovan Herchel) and Slovak Grant Agency VEGA, grant number 1/0125/18 (Ivan Šalitroš).
Acknowledgments
The authors acknowledge Pavla Richterová for performing elemental analysis, Ivana Císařová for X-ray data collection and Ivan Nemec for help with refinement of X-ray data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Decurtins, S.; Gütlich, P.; Kohler, C.P.; Spiering, H.; Hauser, A. Light-induced excited spin state trapping in a transition-metal complex: The hexa-1-propyltetrazole-iron (II) tetrafluoroborate spin-crossover system. Chem. Phys. Lett. 1984, 105, 1–4. [Google Scholar] [CrossRef]
- Hauser, A. Reversibility of light-induced excited spin state trapping in the Fe(ptz)6(BF4)2, and the Zn1−xFex(ptz)6(BF4)2 spin-crossover systems. Chem. Phys. Lett. 1986, 124, 543–548. [Google Scholar] [CrossRef]
- Gutlich, P.; Goodwin, H.A. Spin crossover in transition metal compounds I; Springer: Berlin/Heidelberg, Germany, 2004; pp. 233–235. [Google Scholar] [CrossRef]
- Halcrow, M.A. Spin-Crossover Materials—Properties and Applications; John Wiley & Sons: Chichester, UK, 2013. [Google Scholar]
- Gaspar, A.B.; Ksenofontov, V.; Seredyuk, M.; Gütlich, P. Multifunctionality in spin crossover materials. Coord. Chem. Rev. 2005, 249, 2661–2676. [Google Scholar] [CrossRef]
- Gatteshi, D.; Sessoli, R.; Villain, J. Molecular Nanomegnets; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Gao, S.; Affronte, M.; Baker, M.L.; Blundell, S.; Bogani, L.; Chibotaru, L.F.; Clérac, R.; Cornia, A.; Coulon, C.; Domingo, N. Molecular Nanomagnets and Related Phenomena; Springer: Berlin, Germany, 2015; Volume 164. [Google Scholar]
- Miller, J.S.; Gatteschi, D. Molecule-based magnets themed issue No. 6. Chem. Soc. Rev. 2011, 40, 3065. [Google Scholar] [CrossRef]
- Ababei, R.; Pichon, C.; Roubeau, O.; Li, Y.-G.; Bréfuel, N.; Buisson, L.; Guionneau, P.; Mathonière, C.; Clérac, R. Rational design of a photomagnetic chain: Bridging single-molecule magnets with a spin-crossover complex. J. Am. Chem. Soc. 2013, 135, 14840–14853. [Google Scholar] [CrossRef]
- Mossin, S.; Tran, B.L.; Adhikari, D.; Pink, M.; Heinemann, F.W.; Sutter, J.; Szilagyi, R.K.; Meyer, K.; Mindiola, D.J. A mononuclear Fe(III) single molecule magnet with a 3/2↔5/2 spin crossover. J. Am. Chem. Soc. 2012, 134, 13651–13661. [Google Scholar] [CrossRef]
- Feng, X.; Mathoniere, C.; Jeon, I.R.; Rouzieres, M.; Ozarowski, A.; Aubrey, M.L.; Gonzalez, M.I.; Clerac, R.; Long, J.R. Tristability in a light-actuated single-molecule magnet. J. Am. Chem. Soc. 2013, 135, 15880–15884. [Google Scholar] [CrossRef]
- Urtizberea, A.; Roubeau, O. Switchable slow relaxation of magnetization in the native low temperature phase of a cooperative spin-crossover compound. Chem. Sci. 2017, 8, 2290–2295. [Google Scholar] [CrossRef]
- Mathoniere, C.; Lin, H.J.; Siretanu, D.; Clerac, R.; Smith, J.M. Photoinduced single-molecule magnet properties in a four-coordinate iron(II) spin crossover complex. J. Am. Chem. Soc. 2013, 135, 19083–19086. [Google Scholar] [CrossRef]
- García-López, V.; Orts-Mula, F.J.; Palacios-Corella, M.; Clemente-Juan, J.M.; Clemente-León, M.; Coronado, E. Field-induced slow relaxation of magnetization in a mononuclear Co(II) complex of 2,6-bis(pyrazol-1-yl)pyridine functionalized with a carboxylic acid. Polyhedron 2018, 150, 54–60. [Google Scholar] [CrossRef]
- Goodwin, H.A. Spin crossover in Iron(II) Tris(diimine) and Bis(terimine) systems. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; pp. 59–90. [Google Scholar]
- Halcrow, M.A. Iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridines—A versatile system for spin-crossover research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Cook, L.J.K.; Mohammed, R.; Sherborne, G.; Roberts, T.D.; Alvarez, S.; Halcrow, M.A. Spin state behavior of iron(II)/dipyrazolylpyridine complexes. New insights from crystallographic and solution measurements. Coord. Chem. Rev. 2015, 289, 2–12. [Google Scholar] [CrossRef]
- Bridonneau, N.; Rigamonti, L.; Poneti, G.; Pinkowicz, D.; Forni, A.; Cornia, A. Evidence of crystal packing effects in stabilizing high or low spin states of iron(ii) complexes with functionalized 2,6-bis(pyrazol-1-yl)pyridine ligands. Dalton Trans. 2017, 46, 4075–4085. [Google Scholar] [CrossRef]
- Krüger, C.; Augustín, P.; Nemec, I.; Trávníček, Z.; Oshio, H.; Boča, R.; Renz, F. Spin crossover in Iron(III) complexes with pentadentate schiff base ligands and pseudohalido coligands. Eur. J. Inorg. Chem. 2013, 2013, 902–915. [Google Scholar] [CrossRef]
- Pogány, L.; Brachňaková, B.; Masárová, P.; Moncol, J.; Pavlik, J.; Gál, M.; Mazúr, M.; Herchel, R.; Nemec, I.; Šalitroš, I. Impact of the Schiff base ligand substituents on the solid state and solution properties of eleven iron(iii) complexes. New J. Chem. 2019, 43, 13916–13928. [Google Scholar] [CrossRef]
- Nemec, I.; Svoboda, I.; Herchel, R. Spin crossover in three mononuclear Iron (III) schiff base complexes. Metals 2019, 9, 849. [Google Scholar] [CrossRef]
- El Hajj, F.; Sebki, G.; Patinec, V.; Marchivie, M.; Triki, S.; Handel, H.; Yefsah, S.; Tripier, R.; Gomez-Garcia, C.J.; Coronado, E. Macrocycle-based spin-crossover materials. Inorg. Chem. 2009, 48, 10416–10423. [Google Scholar] [CrossRef]
- Milin, E.; Benaicha, B.; El Hajj, F.; Patinec, V.; Triki, S.; Marchivie, M.; Gómez-García, C.J.; Pillet, S. Magnetic bistability in macrocycle-based FeII Spin-crossover complexes: Counter ion and solvent effects. Eur. J. Inorg. Chem. 2016, 5305–5314. [Google Scholar] [CrossRef]
- Drahoš, B.; Trávníček, Z. Spin crossover Fe(II) complexes of a cross-bridged cyclam derivative. Dalton Trans. 2018, 47, 6134–6145. [Google Scholar] [CrossRef] [PubMed]
- Drahoš, B.; Šalitroš, I.; Herchel, R. First step in preparation of multifunctional spin crossover material based on Fe(II) complex of cyclam-based ligand. Magnetism and DFT studies. Inorg. Chim. Acta 2019, 495, 118921. [Google Scholar] [CrossRef]
- Royal, G.; Dahaoui-Gindrey, V.; Dahaoui, S.; Tabard, A.; Guilard, R.; Pullumbi, P.; Lecomte, C. New synthesis of trans-disubstituted cyclam macrocycles—Elucidation of the disubstitution mechanism on the basis of X-ray data and molecular modeling. Eur. J. Org. Chem. 1998, 1998, 1971–1975. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Perdew, J.P.; Kurth, S.; Zupan, A.; Blaha, P. Accurate density functional with correct formal properties: A step beyond the generalized gradient approximation. Phys. Rev. Lett. 1999, 82, 2544–2547. [Google Scholar] [CrossRef]
- Perdew, J.P.; Tao, J.; Staroverov, V.N.; Scuseria, G.E. Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional. Chem. Phys. 2004, 120, 6898–6911. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A “chain-of-spheres” algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105/1–144105/11. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Boča, R. A Handbook of Magnetochemical Formulae; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Drahoš, B.; Císařová, I.; Laguta, O.; Santana, V.T.; Neugebauer, P.; Herchel, R. Structural, magnetic, redox and theoretical characterization of seven-coordinate first-row transition metal complexes with macrocyclic ligand containing two benzimidazolyl N-pendant arms. Dalton Trans. 2020. [Google Scholar] [CrossRef]
- Boča, R. Zero-field splitting in metal complexes. Coord. Chem. Rev. 2004, 248, 757–815. [Google Scholar] [CrossRef]
- Bar, A.K.; Pichon, C.; Sutter, J.-P. Magnetic anisotropy in two- to eight-coordinated transition–metal complexes: Recent developments in molecular magnetism. Coord. Chem. Rev. 2016, 308, 346–380. [Google Scholar] [CrossRef]
- Cirera, J.; Via-Nadal, M.; Ruiz, E. Benchmarking density functional methods for calculation of state energies of first row spin-crossover molecules. Inorg. Chem. 2018, 57, 14097–14105. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).