Magnetic Properties of Fe(II) Complexes of Cyclam Derivative with One p-Aminobenzyl Pendant Arm


Abstract
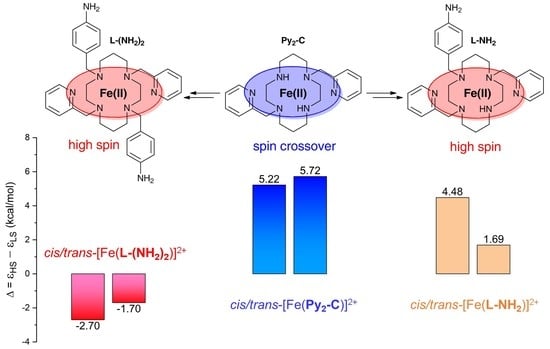
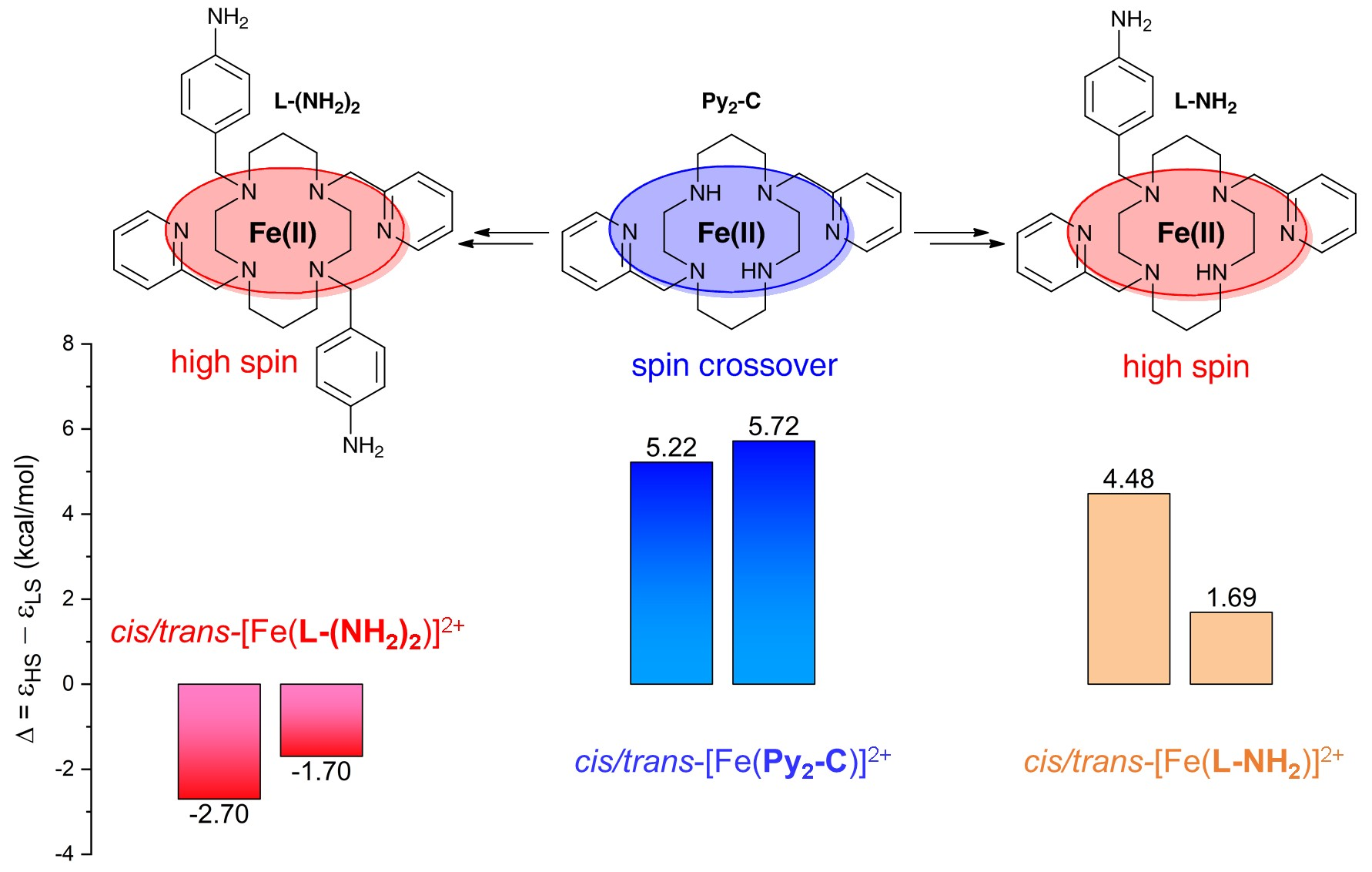{kind=link}
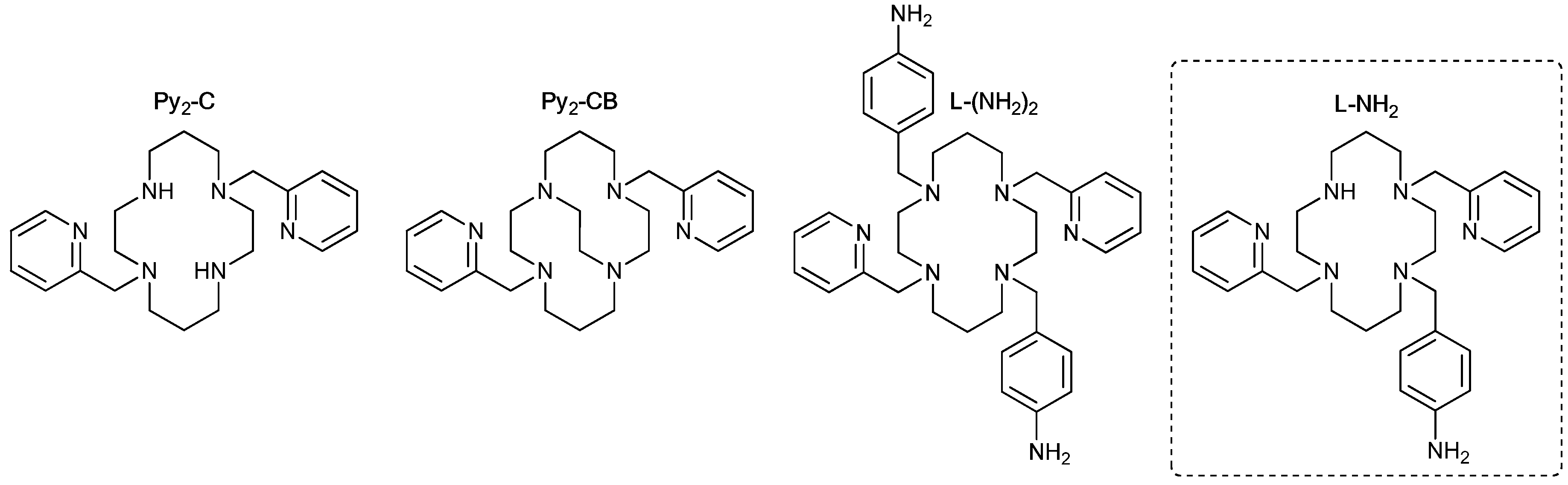{kind=link}
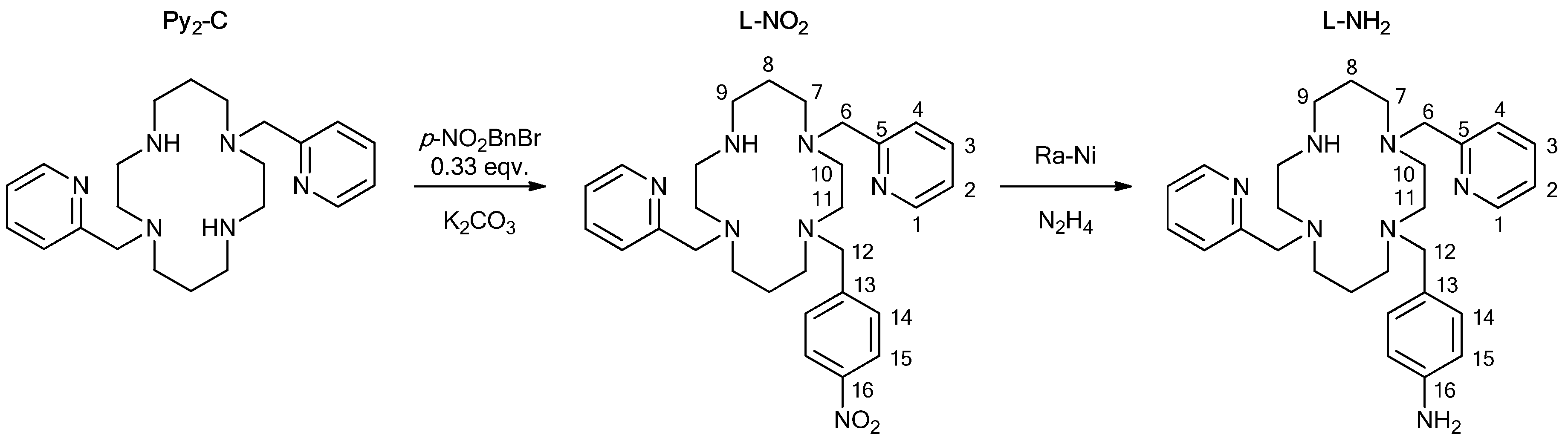{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. Ligand
1-(4-nitrobenzyl)-4,11-bis(pyridine-2-ylmethyl)-1,4,8,11-tetraazacyclotetradecane (L-NO2)
1-(4-aminobenzyl)-4,11-bis(pyridine-2-ylmethyl)-1,4,8,11-tetraazacyclotetradecane (L-NH2)
2.1.2. Complexes 1–3
[Fe(L-NH2)](ClO4)2 (1)
[Fe(L-NH2)]Cl2·6H2O (2)
[Ni(L-NH2)]Cl2·6H2O (3)
2.2. Theoretical Calculations
3. Results and Discussion
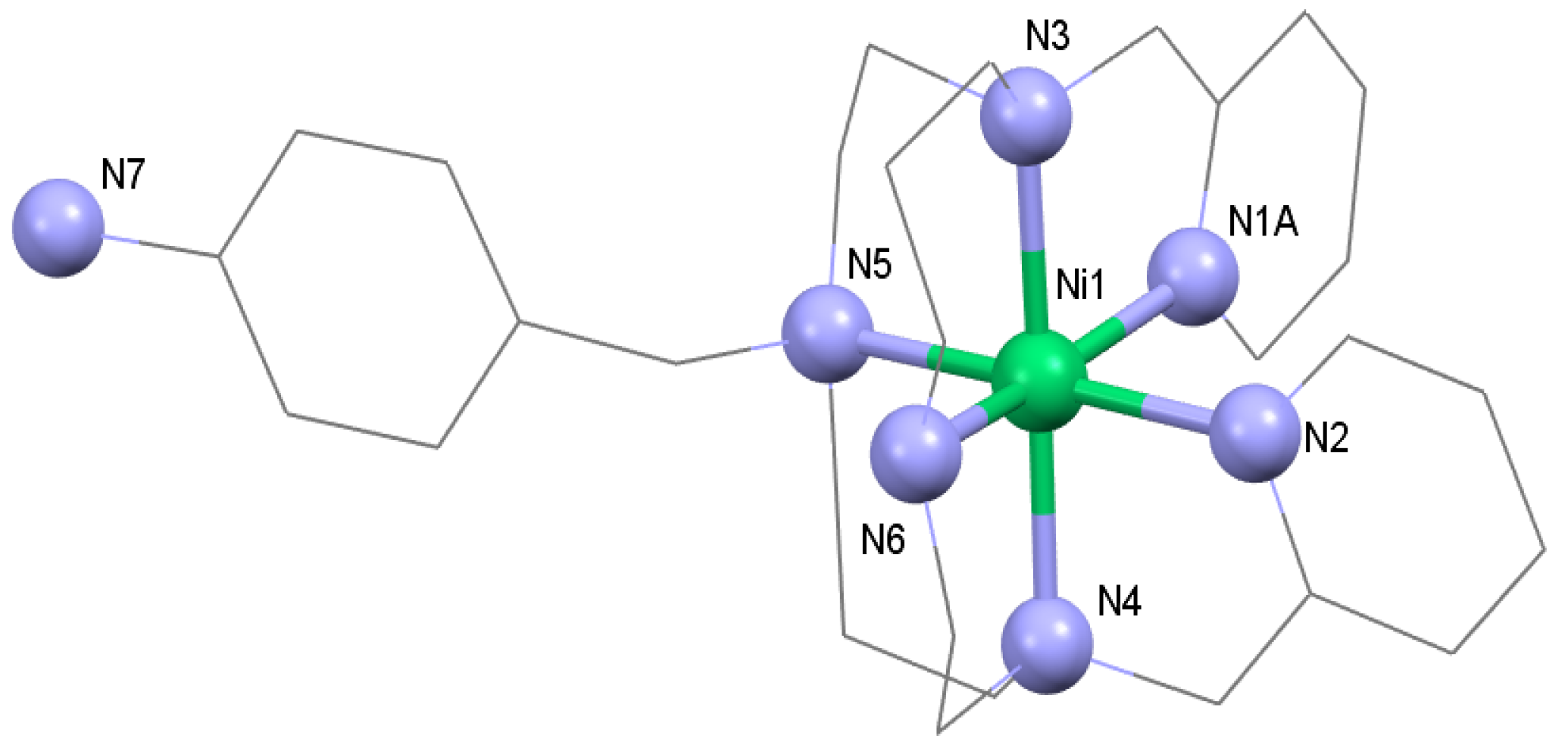
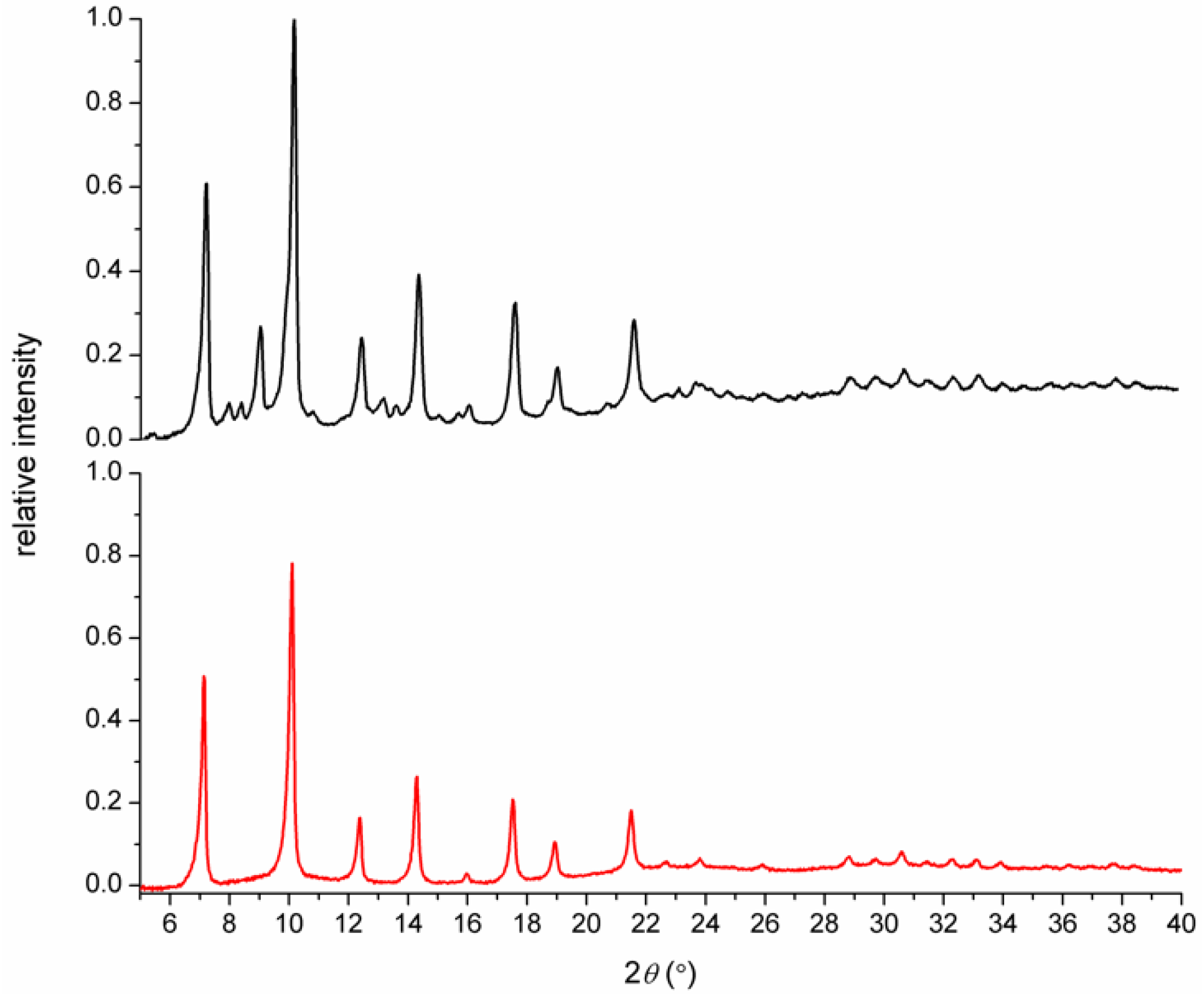
3.1. Syntheses and General Characterizations
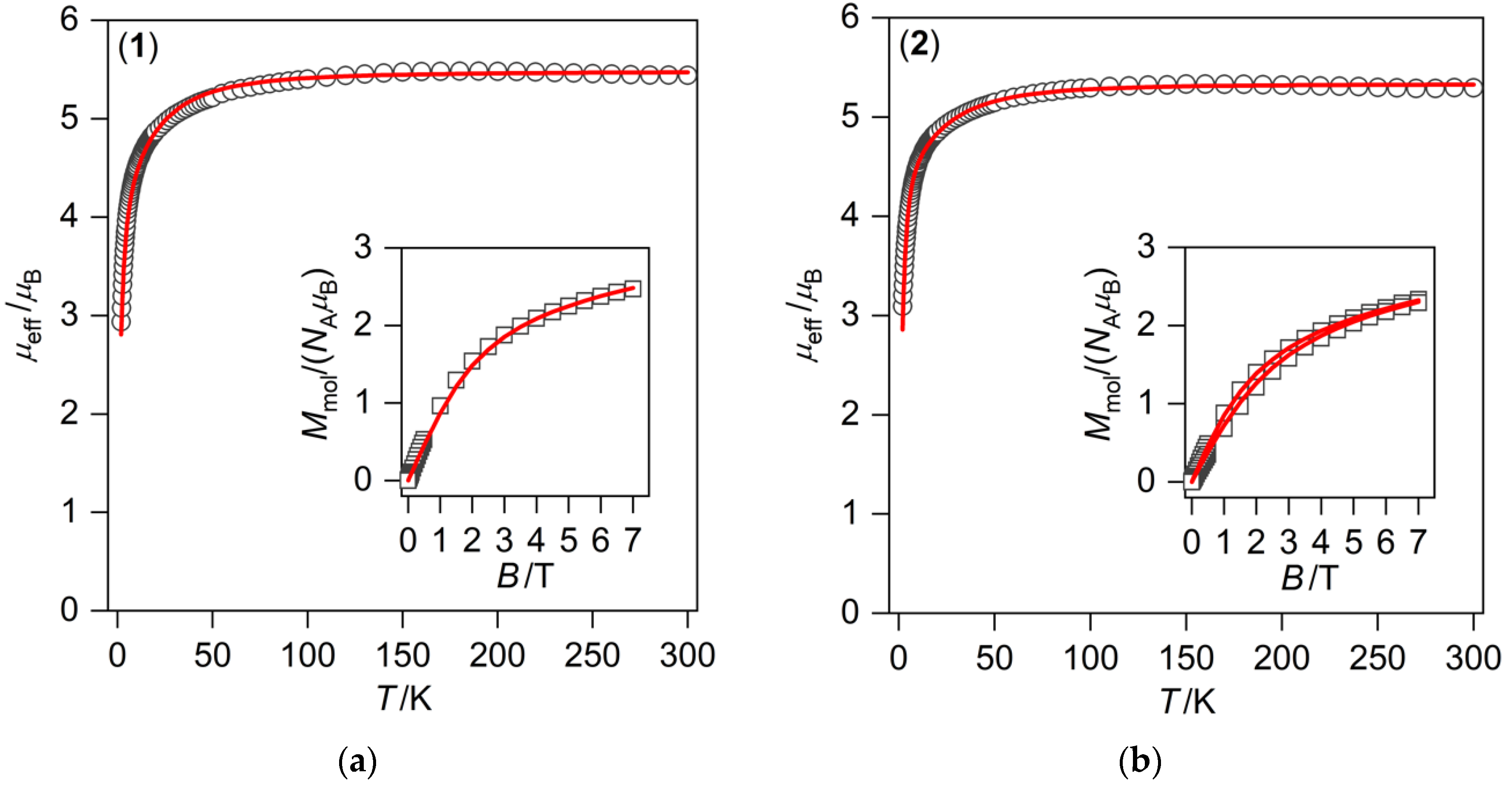
3.2. Magnetic Measurements
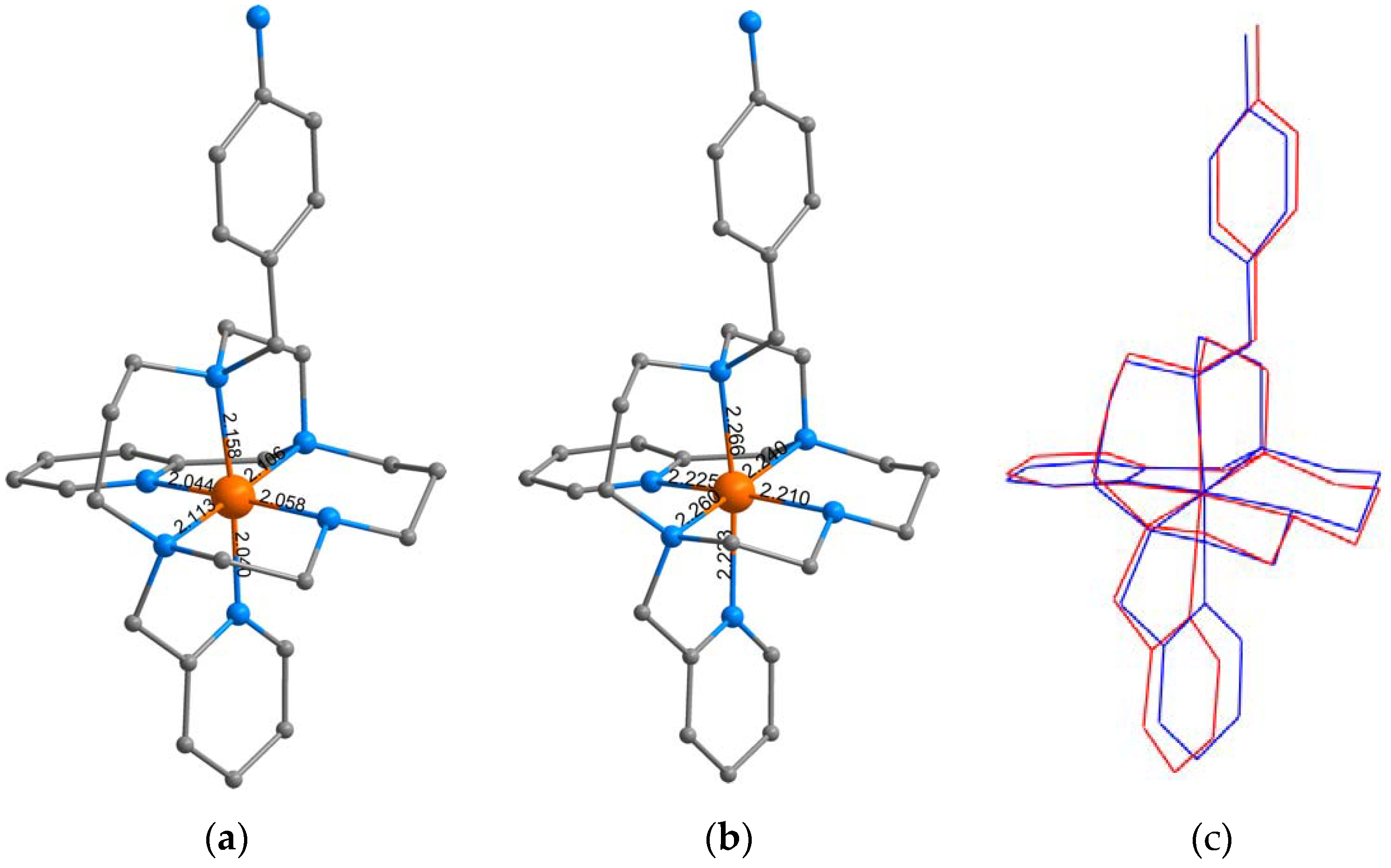
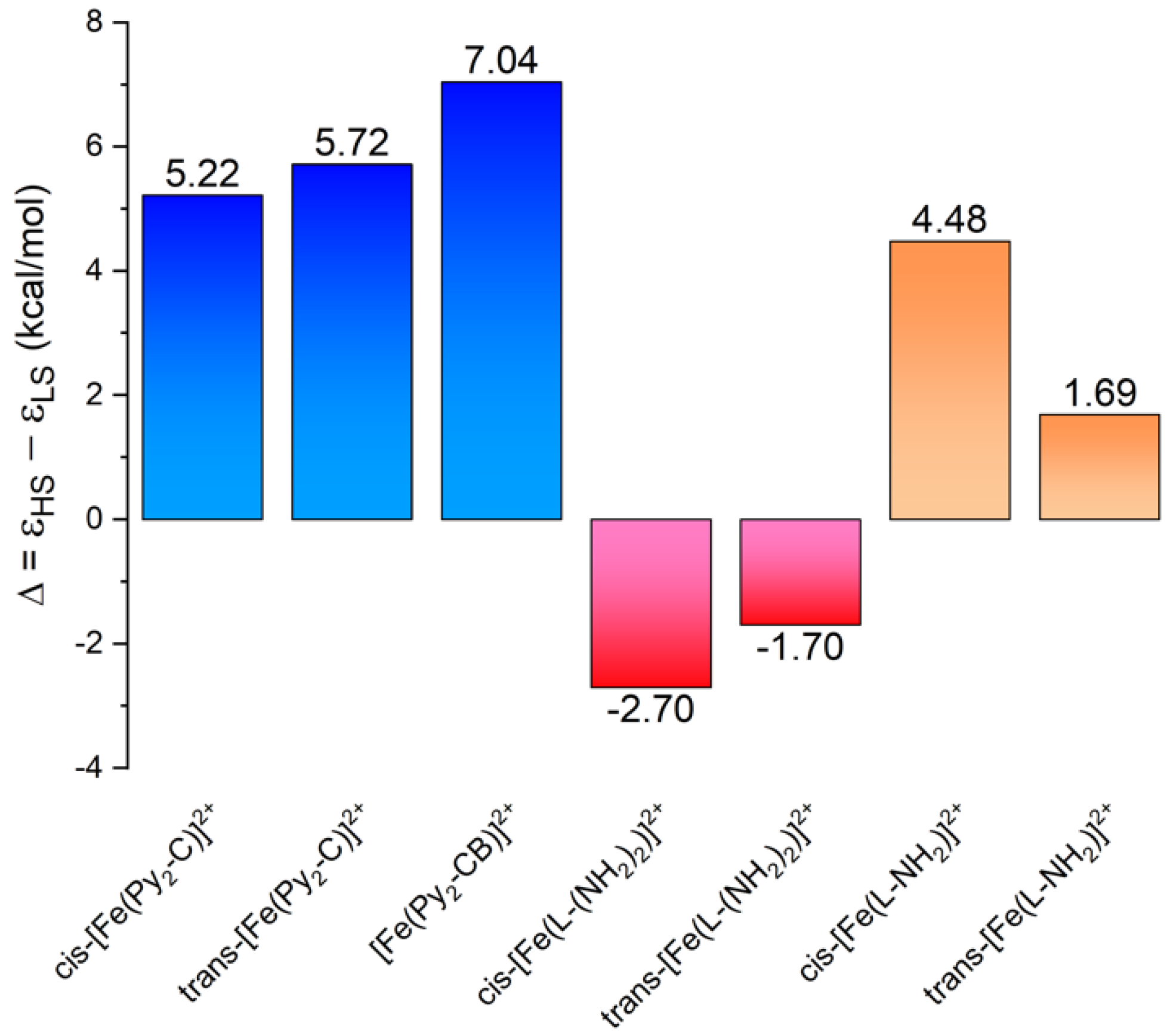
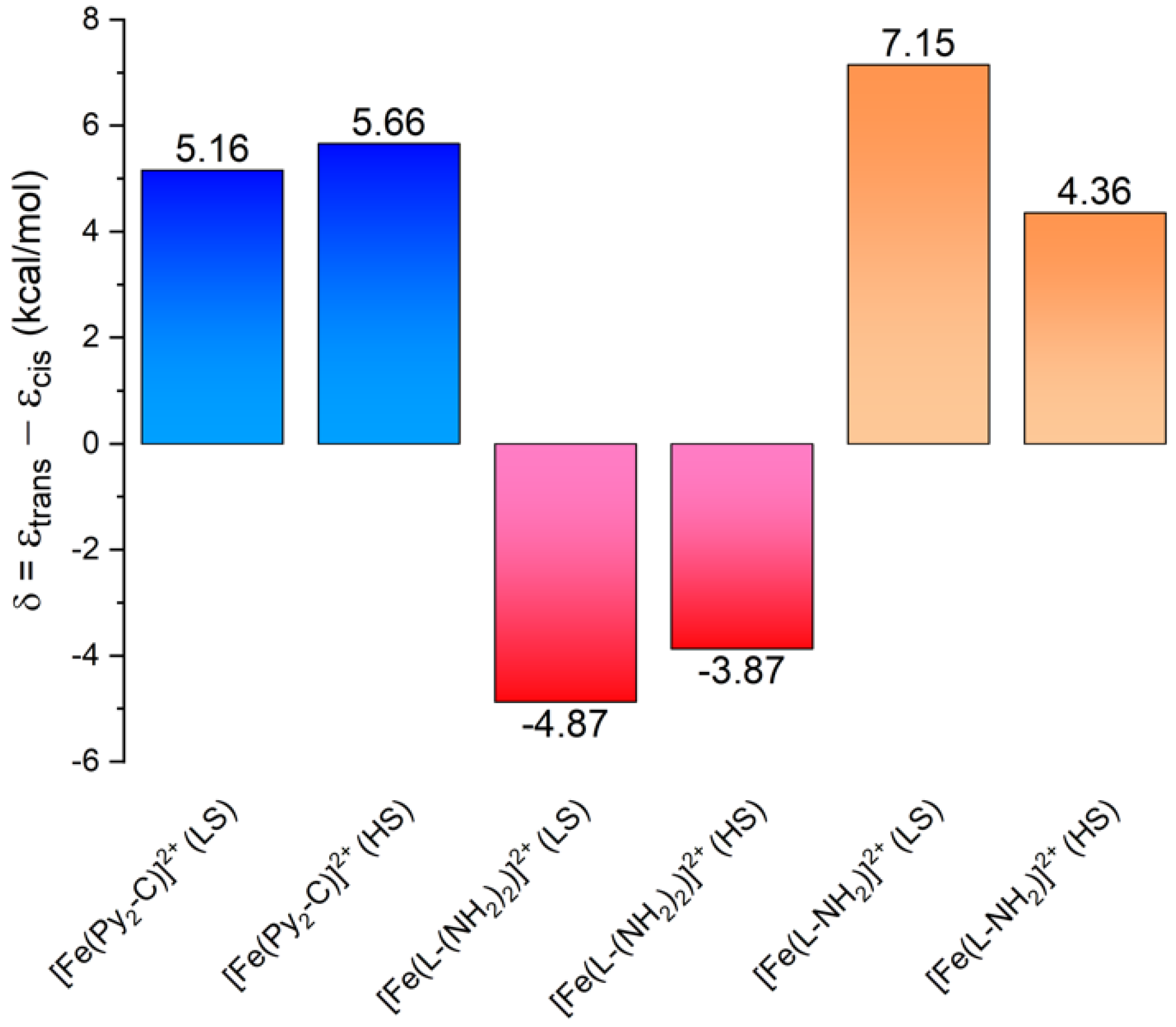
3.3. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Decurtins, S.; Gütlich, P.; Kohler, C.P.; Spiering, H.; Hauser, A. Light-induced excited spin state trapping in a transition-metal complex: The hexa-1-propyltetrazole-iron (II) tetrafluoroborate spin-crossover system. Chem. Phys. Lett. 1984, 105, 1–4. [Google Scholar] [CrossRef]
- Hauser, A. Reversibility of light-induced excited spin state trapping in the Fe(ptz)6(BF4)2, and the Zn1−xFex(ptz)6(BF4)2 spin-crossover systems. Chem. Phys. Lett. 1986, 124, 543–548. [Google Scholar] [CrossRef]
- Gutlich, P.; Goodwin, H.A. Spin crossover in transition metal compounds I; Springer: Berlin/Heidelberg, Germany, 2004; pp. 233–235. [Google Scholar] [CrossRef]
- Halcrow, M.A. Spin-Crossover Materials—Properties and Applications; John Wiley & Sons: Chichester, UK, 2013. [Google Scholar]
- Gaspar, A.B.; Ksenofontov, V.; Seredyuk, M.; Gütlich, P. Multifunctionality in spin crossover materials. Coord. Chem. Rev. 2005, 249, 2661–2676. [Google Scholar] [CrossRef]
- Gatteshi, D.; Sessoli, R.; Villain, J. Molecular Nanomegnets; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Gao, S.; Affronte, M.; Baker, M.L.; Blundell, S.; Bogani, L.; Chibotaru, L.F.; Clérac, R.; Cornia, A.; Coulon, C.; Domingo, N. Molecular Nanomagnets and Related Phenomena; Springer: Berlin, Germany, 2015; Volume 164. [Google Scholar]
- Miller, J.S.; Gatteschi, D. Molecule-based magnets themed issue No. 6. Chem. Soc. Rev. 2011, 40, 3065. [Google Scholar] [CrossRef]
- Ababei, R.; Pichon, C.; Roubeau, O.; Li, Y.-G.; Bréfuel, N.; Buisson, L.; Guionneau, P.; Mathonière, C.; Clérac, R. Rational design of a photomagnetic chain: Bridging single-molecule magnets with a spin-crossover complex. J. Am. Chem. Soc. 2013, 135, 14840–14853. [Google Scholar] [CrossRef]
- Mossin, S.; Tran, B.L.; Adhikari, D.; Pink, M.; Heinemann, F.W.; Sutter, J.; Szilagyi, R.K.; Meyer, K.; Mindiola, D.J. A mononuclear Fe(III) single molecule magnet with a 3/2↔5/2 spin crossover. J. Am. Chem. Soc. 2012, 134, 13651–13661. [Google Scholar] [CrossRef]
- Feng, X.; Mathoniere, C.; Jeon, I.R.; Rouzieres, M.; Ozarowski, A.; Aubrey, M.L.; Gonzalez, M.I.; Clerac, R.; Long, J.R. Tristability in a light-actuated single-molecule magnet. J. Am. Chem. Soc. 2013, 135, 15880–15884. [Google Scholar] [CrossRef]
- Urtizberea, A.; Roubeau, O. Switchable slow relaxation of magnetization in the native low temperature phase of a cooperative spin-crossover compound. Chem. Sci. 2017, 8, 2290–2295. [Google Scholar] [CrossRef]
- Mathoniere, C.; Lin, H.J.; Siretanu, D.; Clerac, R.; Smith, J.M. Photoinduced single-molecule magnet properties in a four-coordinate iron(II) spin crossover complex. J. Am. Chem. Soc. 2013, 135, 19083–19086. [Google Scholar] [CrossRef]
- García-López, V.; Orts-Mula, F.J.; Palacios-Corella, M.; Clemente-Juan, J.M.; Clemente-León, M.; Coronado, E. Field-induced slow relaxation of magnetization in a mononuclear Co(II) complex of 2,6-bis(pyrazol-1-yl)pyridine functionalized with a carboxylic acid. Polyhedron 2018, 150, 54–60. [Google Scholar] [CrossRef]
- Goodwin, H.A. Spin crossover in Iron(II) Tris(diimine) and Bis(terimine) systems. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; pp. 59–90. [Google Scholar]
- Halcrow, M.A. Iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridines—A versatile system for spin-crossover research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Cook, L.J.K.; Mohammed, R.; Sherborne, G.; Roberts, T.D.; Alvarez, S.; Halcrow, M.A. Spin state behavior of iron(II)/dipyrazolylpyridine complexes. New insights from crystallographic and solution measurements. Coord. Chem. Rev. 2015, 289, 2–12. [Google Scholar] [CrossRef]
- Bridonneau, N.; Rigamonti, L.; Poneti, G.; Pinkowicz, D.; Forni, A.; Cornia, A. Evidence of crystal packing effects in stabilizing high or low spin states of iron(ii) complexes with functionalized 2,6-bis(pyrazol-1-yl)pyridine ligands. Dalton Trans. 2017, 46, 4075–4085. [Google Scholar] [CrossRef]
- Krüger, C.; Augustín, P.; Nemec, I.; Trávníček, Z.; Oshio, H.; Boča, R.; Renz, F. Spin crossover in Iron(III) complexes with pentadentate schiff base ligands and pseudohalido coligands. Eur. J. Inorg. Chem. 2013, 2013, 902–915. [Google Scholar] [CrossRef]
- Pogány, L.; Brachňaková, B.; Masárová, P.; Moncol, J.; Pavlik, J.; Gál, M.; Mazúr, M.; Herchel, R.; Nemec, I.; Šalitroš, I. Impact of the Schiff base ligand substituents on the solid state and solution properties of eleven iron(iii) complexes. New J. Chem. 2019, 43, 13916–13928. [Google Scholar] [CrossRef]
- Nemec, I.; Svoboda, I.; Herchel, R. Spin crossover in three mononuclear Iron (III) schiff base complexes. Metals 2019, 9, 849. [Google Scholar] [CrossRef]
- El Hajj, F.; Sebki, G.; Patinec, V.; Marchivie, M.; Triki, S.; Handel, H.; Yefsah, S.; Tripier, R.; Gomez-Garcia, C.J.; Coronado, E. Macrocycle-based spin-crossover materials. Inorg. Chem. 2009, 48, 10416–10423. [Google Scholar] [CrossRef]
- Milin, E.; Benaicha, B.; El Hajj, F.; Patinec, V.; Triki, S.; Marchivie, M.; Gómez-García, C.J.; Pillet, S. Magnetic bistability in macrocycle-based FeII Spin-crossover complexes: Counter ion and solvent effects. Eur. J. Inorg. Chem. 2016, 5305–5314. [Google Scholar] [CrossRef]
- Drahoš, B.; Trávníček, Z. Spin crossover Fe(II) complexes of a cross-bridged cyclam derivative. Dalton Trans. 2018, 47, 6134–6145. [Google Scholar] [CrossRef] [PubMed]
- Drahoš, B.; Šalitroš, I.; Herchel, R. First step in preparation of multifunctional spin crossover material based on Fe(II) complex of cyclam-based ligand. Magnetism and DFT studies. Inorg. Chim. Acta 2019, 495, 118921. [Google Scholar] [CrossRef]
- Royal, G.; Dahaoui-Gindrey, V.; Dahaoui, S.; Tabard, A.; Guilard, R.; Pullumbi, P.; Lecomte, C. New synthesis of trans-disubstituted cyclam macrocycles—Elucidation of the disubstitution mechanism on the basis of X-ray data and molecular modeling. Eur. J. Org. Chem. 1998, 1998, 1971–1975. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Perdew, J.P.; Kurth, S.; Zupan, A.; Blaha, P. Accurate density functional with correct formal properties: A step beyond the generalized gradient approximation. Phys. Rev. Lett. 1999, 82, 2544–2547. [Google Scholar] [CrossRef]
- Perdew, J.P.; Tao, J.; Staroverov, V.N.; Scuseria, G.E. Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional. Chem. Phys. 2004, 120, 6898–6911. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A “chain-of-spheres” algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105/1–144105/11. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Boča, R. A Handbook of Magnetochemical Formulae; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Drahoš, B.; Císařová, I.; Laguta, O.; Santana, V.T.; Neugebauer, P.; Herchel, R. Structural, magnetic, redox and theoretical characterization of seven-coordinate first-row transition metal complexes with macrocyclic ligand containing two benzimidazolyl N-pendant arms. Dalton Trans. 2020. [Google Scholar] [CrossRef]
- Boča, R. Zero-field splitting in metal complexes. Coord. Chem. Rev. 2004, 248, 757–815. [Google Scholar] [CrossRef]
- Bar, A.K.; Pichon, C.; Sutter, J.-P. Magnetic anisotropy in two- to eight-coordinated transition–metal complexes: Recent developments in molecular magnetism. Coord. Chem. Rev. 2016, 308, 346–380. [Google Scholar] [CrossRef]
- Cirera, J.; Via-Nadal, M.; Ruiz, E. Benchmarking density functional methods for calculation of state energies of first row spin-crossover molecules. Inorg. Chem. 2018, 57, 14097–14105. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drahoš, B.; Antal, P.; Šalitroš, I.; Herchel, R. Magnetic Properties of Fe(II) Complexes of Cyclam Derivative with One p-Aminobenzyl Pendant Arm. Metals 2020, 10, 366. https://doi.org/10.3390/met10030366
Drahoš B, Antal P, Šalitroš I, Herchel R. Magnetic Properties of Fe(II) Complexes of Cyclam Derivative with One p-Aminobenzyl Pendant Arm. Metals. 2020; 10(3):366. https://doi.org/10.3390/met10030366
Chicago/Turabian StyleDrahoš, Bohuslav, Peter Antal, Ivan Šalitroš, and Radovan Herchel. 2020. "Magnetic Properties of Fe(II) Complexes of Cyclam Derivative with One p-Aminobenzyl Pendant Arm" Metals 10, no. 3: 366. https://doi.org/10.3390/met10030366
APA StyleDrahoš, B., Antal, P., Šalitroš, I., & Herchel, R. (2020). Magnetic Properties of Fe(II) Complexes of Cyclam Derivative with One p-Aminobenzyl Pendant Arm. Metals, 10(3), 366. https://doi.org/10.3390/met10030366