Prediction of Equilibrium Phase, Stability and Stress-Strain Properties in Co-Cr-Fe-Ni-Al High Entropy Alloys Using Artificial Neural Networks


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
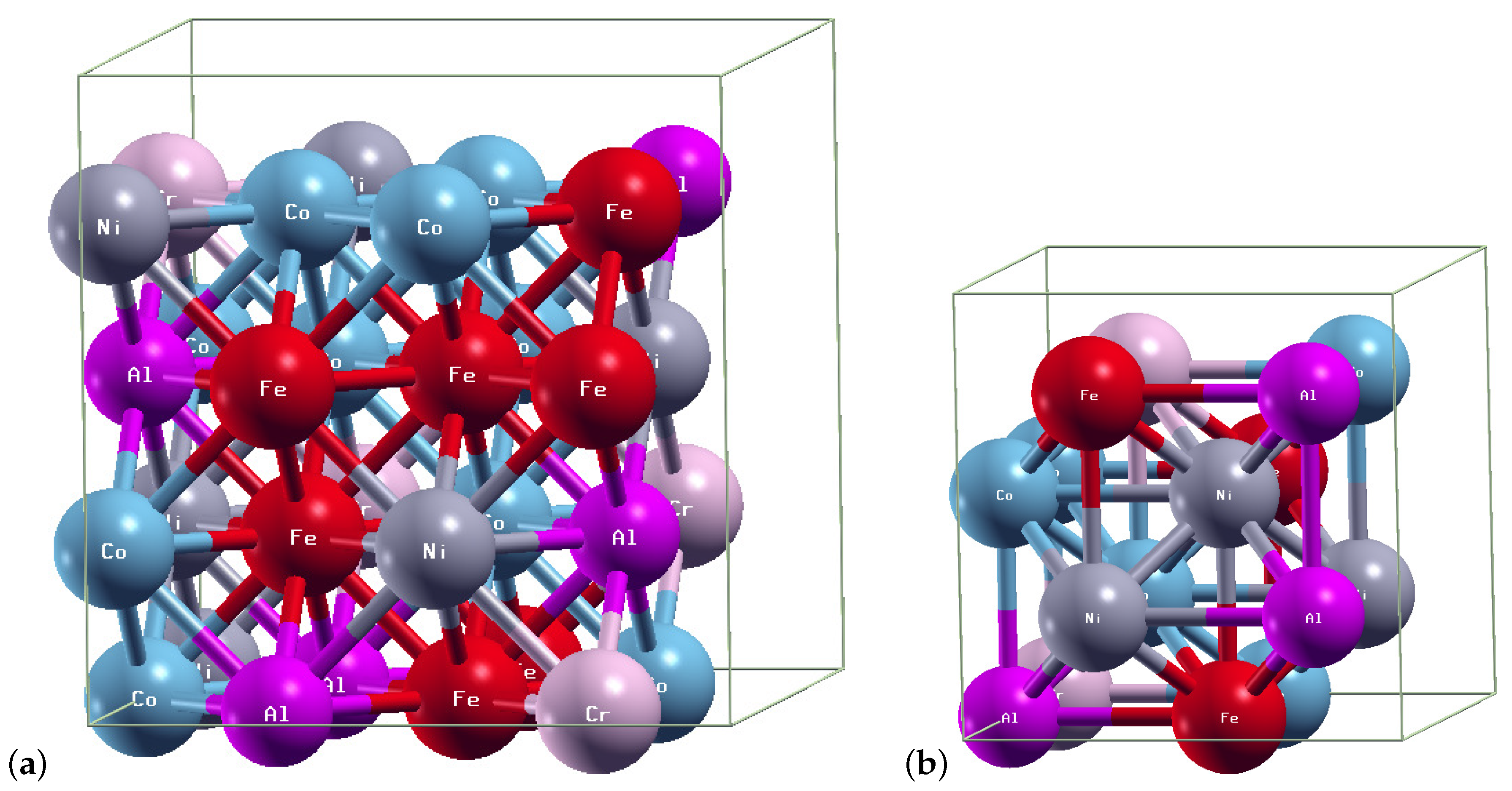
2. Models and Methods
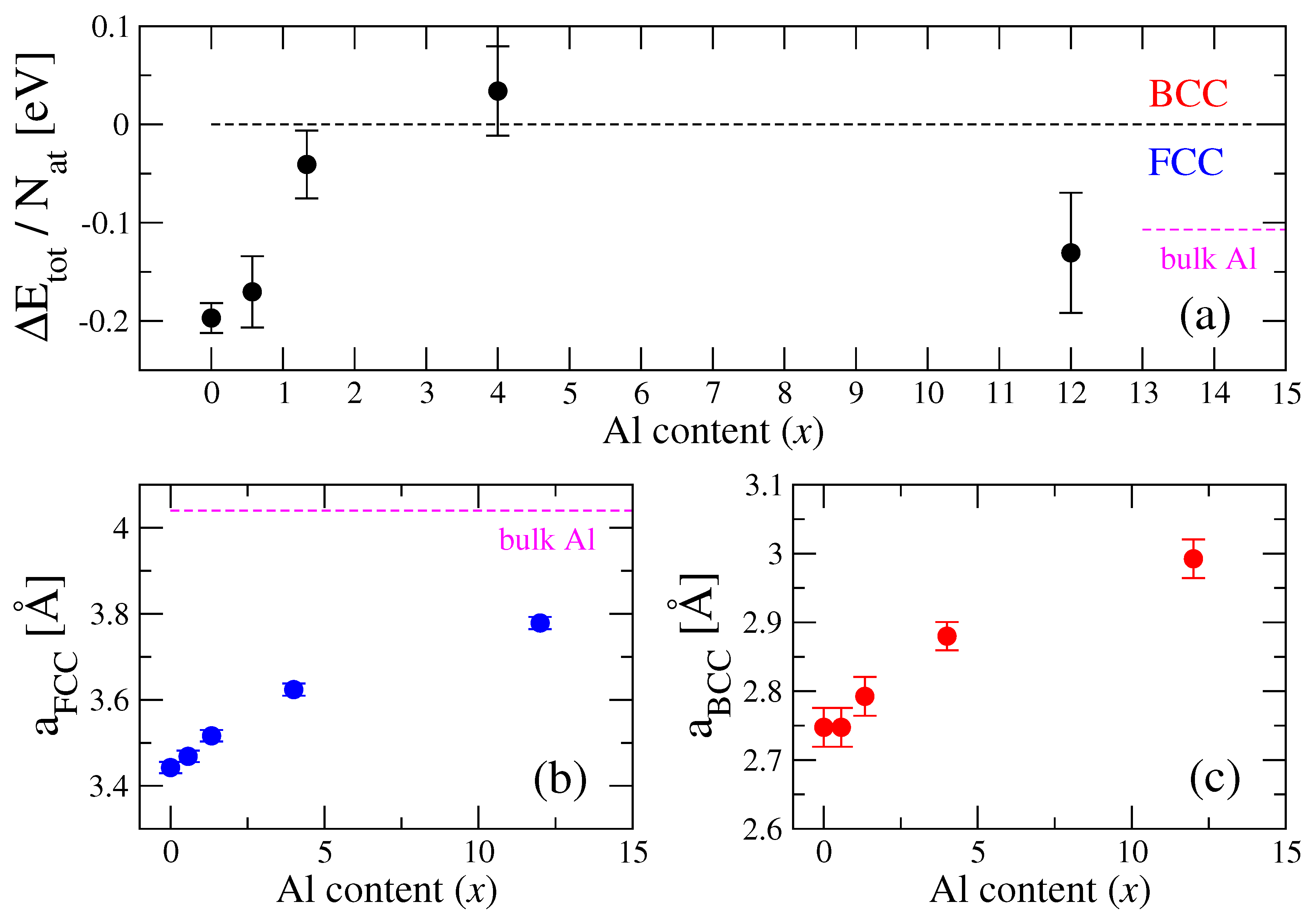
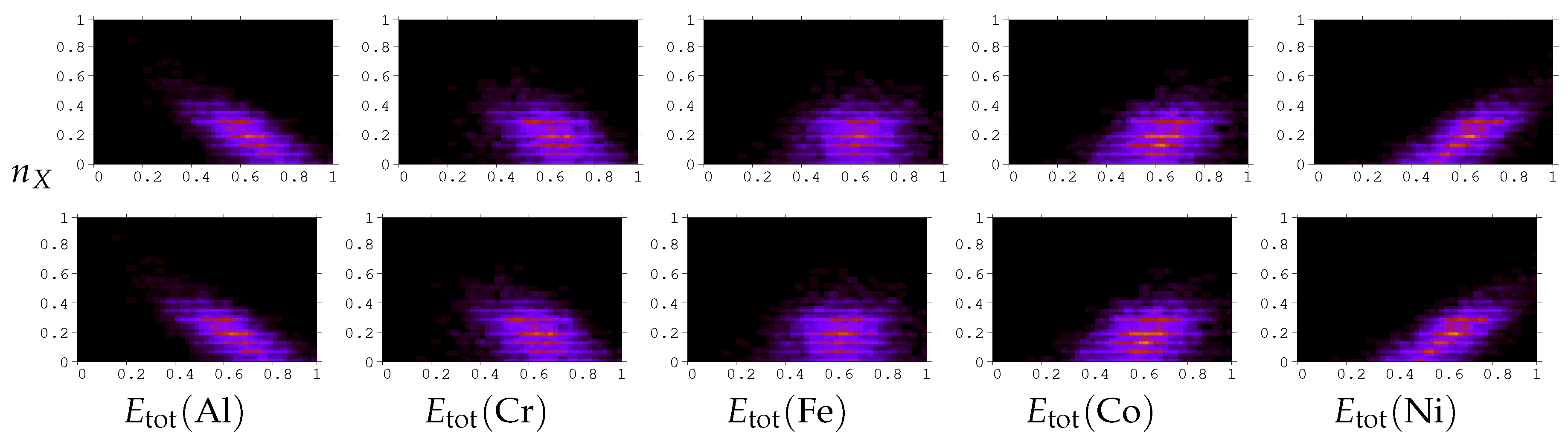
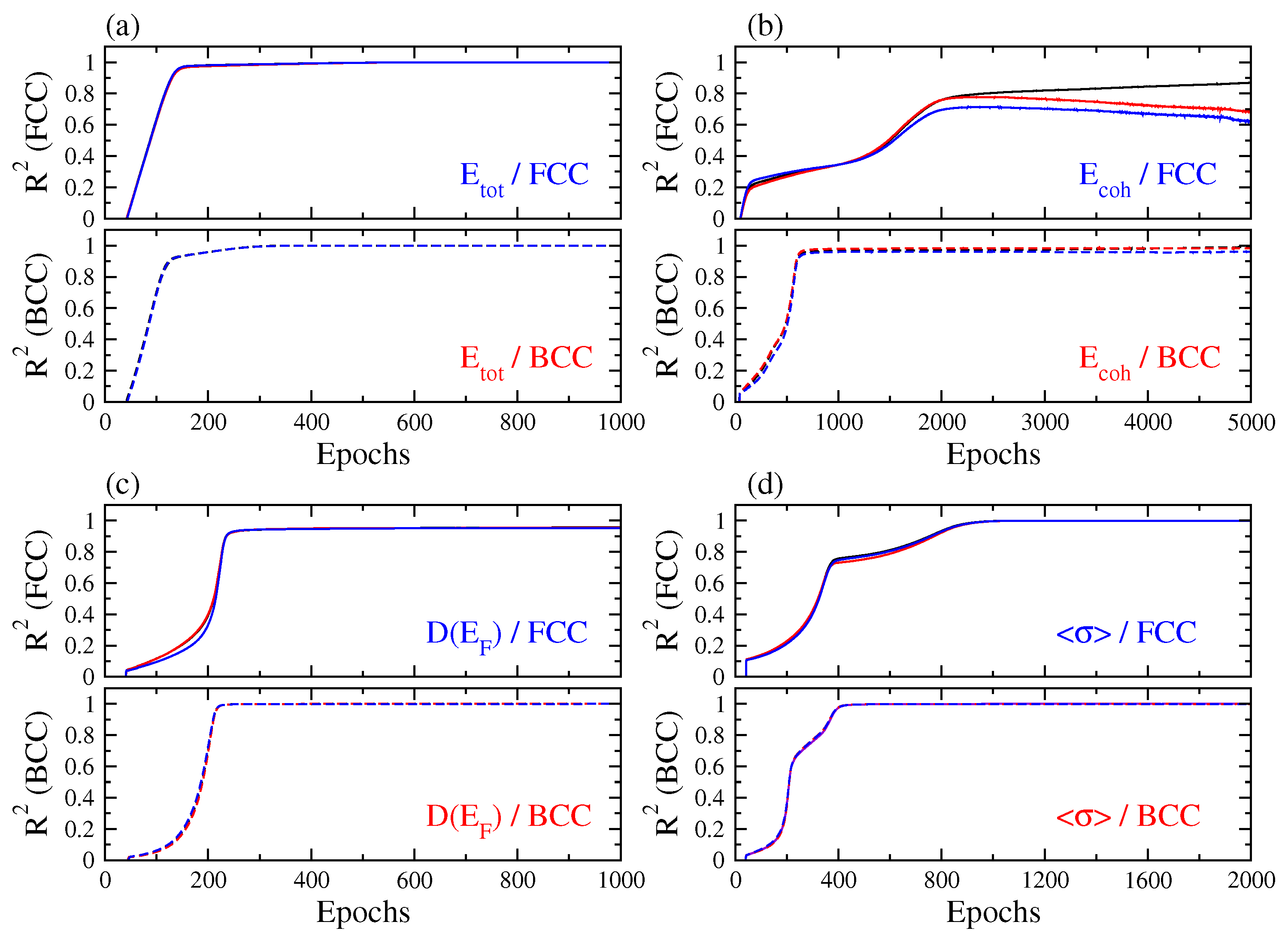
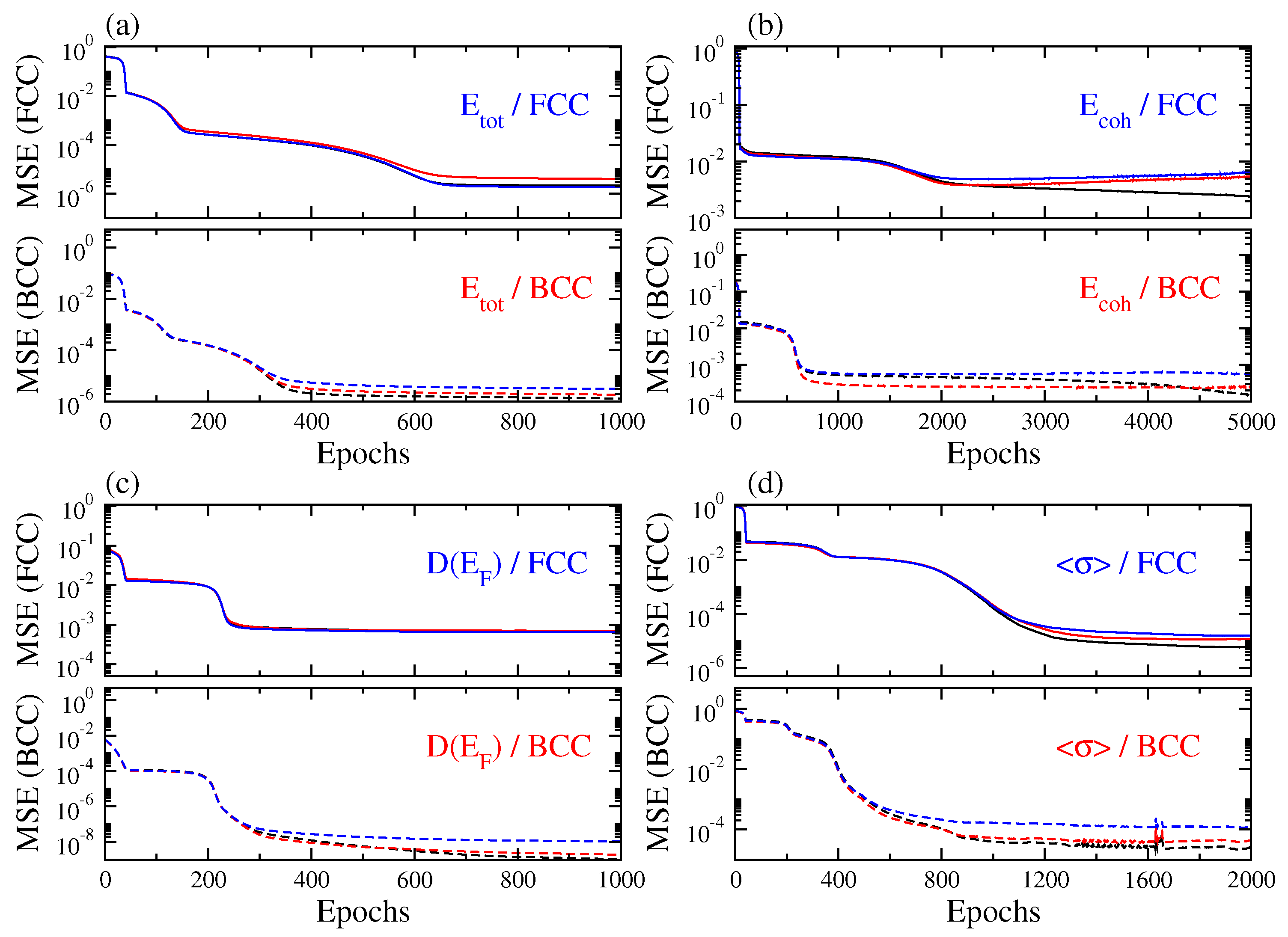
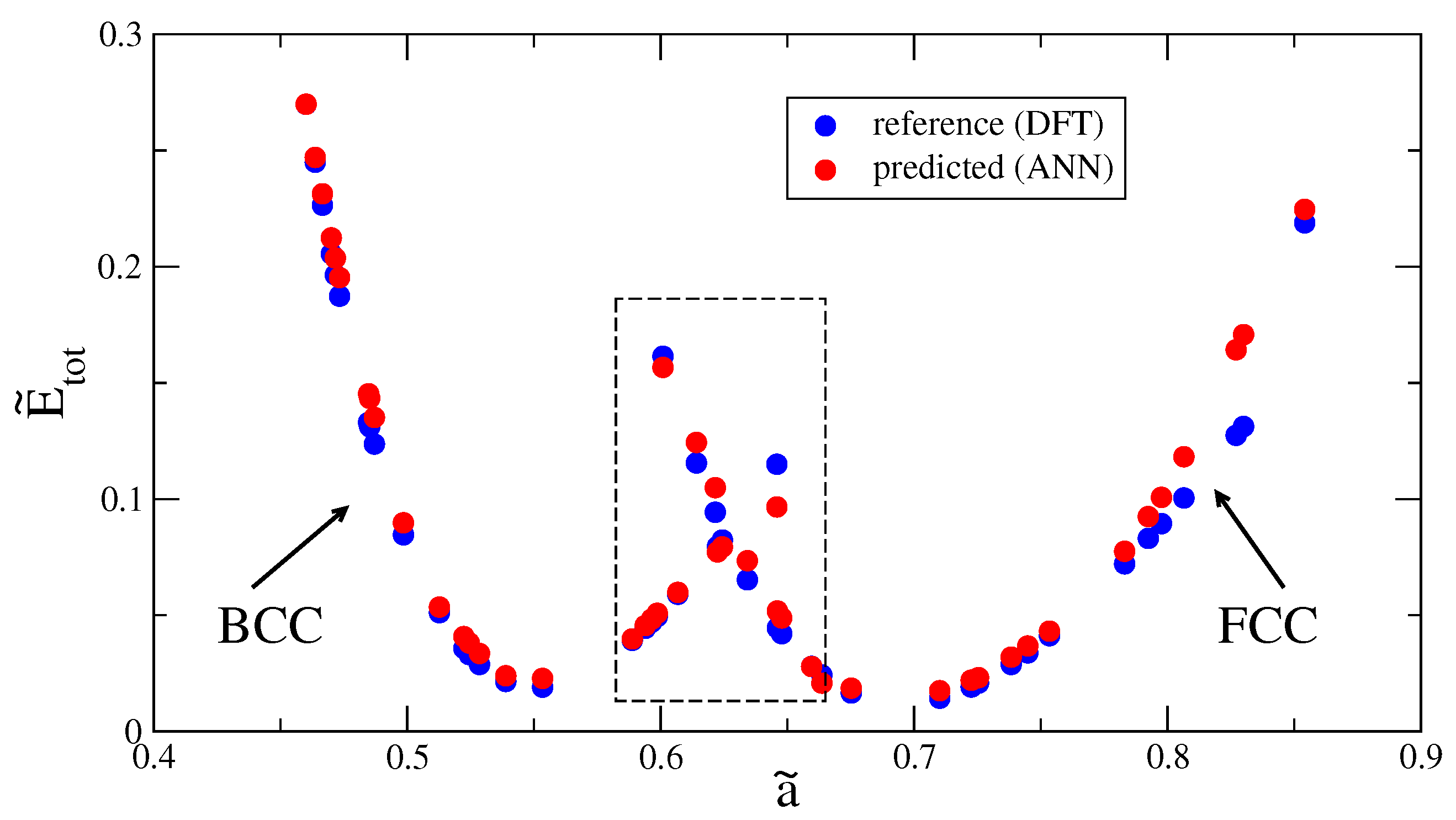
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gorsse, S.; Couzinié, J.P.; Miracle, D.B. From high-entropy alloys to complex concentrated alloys. Comptes Rendus Phys. 2018, 19, 721–736. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, X.; Wang, W.; Liu, B.; Lv, Y.; Yang, W.; Xu, D.; Liu, Y. A review on fundamental of high entropy alloys with promising high–temperature properties. J. Alloys Compd. 2018, 760, 15–30. [Google Scholar] [CrossRef]
- George, E.P.; Raabe, D.; Ritchie, R.O. High-entropy alloys. Nat. Rev. Mater. 2019, 4, 515–534. [Google Scholar] [CrossRef]
- Qiu, S.; Miao, N.; Zhou, J.; Guo, Z.; Sun, Z. Strengthening mechanism of aluminum on elastic properties of NbVTiZr high-entropy alloys. Intermetallics 2018, 92, 7–14. [Google Scholar] [CrossRef]
- Wu, J.M.; Lin, S.J.; Yeh, J.W.; Chen, S.K.; Huang, Y.S.; Chen, H.C. Adhesive wear behavior of AlxCoCrCuFeNi high-entropy alloys as a function of aluminum content. Wear 2006, 261, 513–519. [Google Scholar] [CrossRef]
- Chuang, M.H.; Tsai, M.H.; Wang, W.R.; Lin, S.J.; Yeh, J.W. Microstructure and wear behavior of AlxCo1.5CrFeNi1.5Tiy high-entropy alloys. Acta Mater. 2011, 59, 6308–6317. [Google Scholar] [CrossRef]
- Qiu, Y.; Thomas, S.; Gibson, M.A.; Fraser, H.L.; Birbilis, N. Corrosion of high entropy alloys. Npj Mater. Degrad. 2017, 1, 15. [Google Scholar] [CrossRef]
- Huang, P.K.; Yeh, J.W.; Shun, T.T.; Chen, S.K. Multi-Principal-Element Alloys with Improved Oxidation and Wear Resistance for Thermal Spray Coating. Adv. Eng. Mater. 2004, 6, 74–78. [Google Scholar] [CrossRef]
- Kumar, N.K.; Li, C.; Leonard, K.; Bei, H.; Zinkle, S. Microstructural stability and mechanical behavior of FeNiMnCr high entropy alloy under ion irradiation. Acta Mater. 2016, 113, 230–244. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, L.; Cheng, H.; Zhang, H.; Ma, M.; Li, Y.; Li, G.; Liaw, P.; Liu, R. The high-entropy alloys with high hardness and soft magnetic property prepared by mechanical alloying and high-pressure sintering. Intermetallics 2016, 70, 82–87. [Google Scholar] [CrossRef]
- Sriharitha, R.; Murty, B.; Kottada, R.S. Alloying, thermal stability and strengthening in spark plasma sintered AlxCoCrCuFeNi high entropy alloys. J. Alloys Compd. 2014, 583, 419–426. [Google Scholar] [CrossRef]
- Cantor, B.; Chang, I.; Knight, P.; Vincent, A. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375–377, 213–218. [Google Scholar] [CrossRef]
- Yeh, J.W.; Chen, S.K.; Lin, S.J.; Gan, J.Y.; Chin, T.S.; Shun, T.T.; Tsau, C.H.; Chang, S.Y. Nanostructured High-Entropy Alloys with Multiple Principal Elements: Novel Alloy Design Concepts and Outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Senkov, O.; Wilks, G.; Miracle, D.; Chuang, C.; Liaw, P. Refractory high-entropy alloys. Intermetallics 2010, 18, 1758–1765. [Google Scholar] [CrossRef]
- Mishra, R.K.; Shahi, R.R. Phase evolution and magnetic characteristics of TiFeNiCr and TiFeNiCrM (M=Mn, Co) high entropy alloys. J. Magn. Magn. Mater. 2017, 442, 218–223. [Google Scholar] [CrossRef]
- Yuan, Y.; Wu, Y.; Tong, X.; Zhang, H.; Wang, H.; Liu, X.; Ma, L.; Suo, H.; Lu, Z. Rare-earth high-entropy alloys with giant magnetocaloric effect. Acta Mater. 2017, 125, 481–489. [Google Scholar] [CrossRef]
- Sarlar, K.; Tekgül, A.; Kucuk, I. Magnetocaloric properties in a FeNiGaMnSi high entropy alloy. Curr. Appl. Phys. 2020, 20, 18–22. [Google Scholar] [CrossRef]
- He, F.; Wang, Z.; Wu, Q.; Niu, S.; Li, J.; Wang, J.; Liu, C. Solid solution island of the Co-Cr-Fe-Ni high entropy alloy system. Scr. Mater. 2017, 131, 42–46. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, S.; Qiao, J.; Wang, Z.; Gao, M.; Jiao, Z.; Yang, H.; Zhang, Y. Superior high tensile elongation of a single-crystal CoCrFeNiAl0.3 high-entropy alloy by Bridgman solidification. Intermetallics 2014, 54, 104–109. [Google Scholar] [CrossRef]
- He, J.; Wang, H.; Huang, H.; Xu, X.; Chen, M.; Wu, Y.; Liu, X.; Nieh, T.; An, K.; Lu, Z. A precipitation-hardened high-entropy alloy with outstanding tensile properties. Acta Mater. 2016, 102, 187–196. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, Y. Effect of Nb addition on the microstructure and properties of AlCoCrFeNi high-entropy alloy. Mater. Sci. Eng. A 2012, 532, 480–486. [Google Scholar] [CrossRef]
- Prasad, H.; Singh, S.; Panigrahi, B.B. Mechanical activated synthesis of alumina dispersed FeNiCoCrAlMn high entropy alloy. J. Alloys Compd. 2017, 692, 720–726. [Google Scholar] [CrossRef]
- Chou, H.P.; Chang, Y.S.; Chen, S.K.; Yeh, J.W. Microstructure, thermophysical and electrical properties in AlxCoCrFeNi (0 ≤ x ≤ 2) high-entropy alloys. Mater. Sci. Eng. B 2009, 163, 184–189. [Google Scholar] [CrossRef]
- Kao, Y.F.; Chen, T.J.; Chen, S.K.; Yeh, J.W. Microstructure and mechanical property of as-cast, -homogenized, and -deformed AlxCoCrFeNi (0 ≤ x ≤ 2) high-entropy alloys. J. Alloys Compd. 2009, 488, 57–64. [Google Scholar] [CrossRef]
- Wang, W.R.; Wang, W.L.; Wang, S.C.; Tsai, Y.C.; Lai, C.H.; Yeh, J.W. Effects of Al addition on the microstructure and mechanical property of AlxCoCrFeNi high-entropy alloys. Intermetallics 2012, 26, 44–51. [Google Scholar] [CrossRef]
- Tian, F.; Delczeg, L.; Chen, N.; Varga, L.K.; Shen, J.; Vitos, L. Structural stability of NiCoFeCrAlx high-entropy alloy from ab initio theory. Phys. Rev. B 2013, 88, 085128. [Google Scholar] [CrossRef]
- Widom, M. Modeling the structure and thermodynamics of high-entropy alloys. J. Mater. Res. 2018, 33, 2881–2898. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhou, Y.; He, Q.; Ding, Z.; Li, F.; Yang, Y. Machine learning guided appraisal and exploration of phase design for high entropy alloys. Npj Comput. Mater. 2019, 5, 128. [Google Scholar] [CrossRef]
- Huang, W.; Martin, P.; Zhuang, H.L. Machine-learning phase prediction of high-entropy alloys. Acta Mater. 2019, 169, 225–236. [Google Scholar] [CrossRef]
- Chang, Y.J.; Jui, C.Y.; Lee, W.J.; Yeh, A.C. Prediction of the Composition and Hardness of High-Entropy Alloys by Machine Learning. JOM 2019, 71, 3433–3442. [Google Scholar] [CrossRef]
- Wen, C.; Zhang, Y.; Wang, C.; Xue, D.; Bai, Y.; Antonov, S.; Dai, L.; Lookman, T.; Su, Y. Machine learning assisted design of high entropy alloys with desired property. Acta Mater. 2019, 170, 109–117. [Google Scholar] [CrossRef]
- Gao, M.C.; Niu, C.; Jiang, C.; Irving, D.L. Applications of Special Quasi-random Structures to High-Entropy Alloys. In High-Entropy Alloys: Fundamentals and Applications; Gao, M.C., Yeh, J.W., Liaw, P.K., Zhang, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 333–368. [Google Scholar]
- Soler, J.M.; Artacho, E.; Gale, J.D.; Garcia, A.; Junquera, J.; Ordejon, P.; Sanchez-Portal, D. The SIESTA method for ab initio order- N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Li, S. Cohesive Energy: The Intrinsic Dominant of Thermal Stability and Structural Evolution in Sn from Size Scales of Bulk to Dimer. J. Phys. Chem. C 2009, 113, 14207–14212. [Google Scholar] [CrossRef]
- TensorFlow. Available online: https://www.tensorflow.org/ (accessed on 1 June 2020).
- Keras. Available online: https://keras.io/ (accessed on 1 June 2020).
- Nemnes, G.A.; Mitran, T.L.; Manolescu, A. Gap prediction in hybrid graphene-hexagonal boron nitride nanoflakes using artificial neural networks. J. Nanometer. 2019, 2019, 6960787. [Google Scholar]
- Mitran, T.; Nemnes, G. Theory and Simulation in Physics for Materials Applications. In Prediction of Energy Gaps in Graphene-Hexagonal Boron Nitride Nanoflakes Using Artificial Neural Networks; Springer International Publishing: Berlin, Germany, 2020; pp. 197–209. [Google Scholar]
- Zaddach, A.J.; Niu, C.; Koch, C.C.; Irving, D.L. Mechanical Properties and Stacking Fault Energies of NiFeCrCoMn High-Entropy Alloy. JOM 2013, 65, 1780–1789. [Google Scholar] [CrossRef]
- Tang, Z.; Senkov, O.N.; Parish, C.M.; Zhang, C.; Zhang, F.; Santodonato, L.J.; Wang, G.; Zhao, G.; Yang, F.; Liaw, P.K. Tensile ductility of an AlCoCrFeNi multi-phase high-entropy alloy through hot isostatic pressing (HIP) and homogenization. Mater. Sci. Eng. A 2015, 647, 229–240. [Google Scholar] [CrossRef]
- Li, D.; Li, C.; Feng, T.; Zhang, Y.; Sha, G.; Lewandowski, J.J.; Liaw, P.K.; Zhang, Y. High-entropy Al0.3CoCrFeNi alloy fibers with high tensile strength and ductility at ambient and cryogenic temperatures. Acta Mater. 2017, 123, 285–294. [Google Scholar] [CrossRef]
- Wu, Z.; Bei, H.; Otto, F.; Pharr, G.; George, E. Recovery, recrystallization, grain growth and phase stability of a family of FCC-structured multi-component equiatomic solid solution alloys. Intermetallics 2014, 46, 131–140. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, T.; Qiao, J.; Ma, S.; Zhao, D.; Lu, P.; Wang, Z. Mechanical properties and deformation behavior of dual-phase Al0.6CoCrFeNi high-entropy alloys with heterogeneous structure at room and cryogenic temperatures. J. Alloys Compd. 2020, 816, 152663. [Google Scholar] [CrossRef]
- Mehta, A.; Sohn, Y. Interdiffusion, Solubility Limit, and Role of Entropy in FCC Al-Co-Cr-Fe-Ni Alloys. Metall. Mater. Trans. A 2020, 51, 3142–3153. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipoiu, N.; Nemnes, G.A. Prediction of Equilibrium Phase, Stability and Stress-Strain Properties in Co-Cr-Fe-Ni-Al High Entropy Alloys Using Artificial Neural Networks. Metals 2020, 10, 1569. https://doi.org/10.3390/met10121569
Filipoiu N, Nemnes GA. Prediction of Equilibrium Phase, Stability and Stress-Strain Properties in Co-Cr-Fe-Ni-Al High Entropy Alloys Using Artificial Neural Networks. Metals. 2020; 10(12):1569. https://doi.org/10.3390/met10121569
Chicago/Turabian StyleFilipoiu, Nicolae, and George Alexandru Nemnes. 2020. "Prediction of Equilibrium Phase, Stability and Stress-Strain Properties in Co-Cr-Fe-Ni-Al High Entropy Alloys Using Artificial Neural Networks" Metals 10, no. 12: 1569. https://doi.org/10.3390/met10121569
APA StyleFilipoiu, N., & Nemnes, G. A. (2020). Prediction of Equilibrium Phase, Stability and Stress-Strain Properties in Co-Cr-Fe-Ni-Al High Entropy Alloys Using Artificial Neural Networks. Metals, 10(12), 1569. https://doi.org/10.3390/met10121569