First Principles Study of Bonding Mechanisms at the TiAl/TiO2 Interface

 ,
, 
Abstract
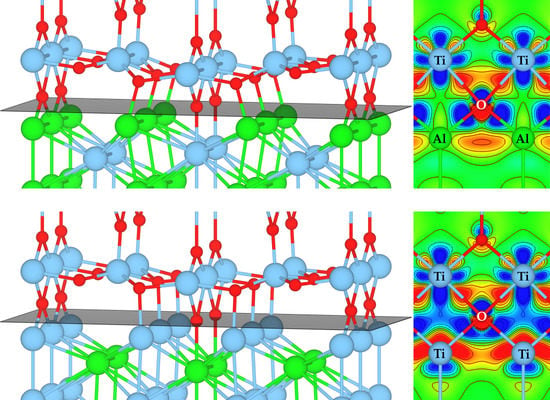
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Surface Energy
3.2. Work of Separation
3.3. Atomic and Electronic Factors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Gao, W. High temperature corrosion of intermetallics. In Intermetallics Research Progress; Berdovsky, N., Ed.; Nova Science: New York, NY, USA, 2008; pp. 1–64. [Google Scholar]
- Dai, J.; Zhu, J.; Chen, C.; Weng, F. High temperature oxidation behavior and research status of modifications on improving high temperature oxidation resistance of titanium alloys and titanium aluminides: A review. J. Alloys Compd. 2016, 685, 784–798. [Google Scholar] [CrossRef]
- Shanabarger, M.R. Comparative study of the initial oxidation behavior of a series of titanium–aluminum alloys. Appl. Surf. Sci. 1998, 134, 179–186. [Google Scholar] [CrossRef]
- Maurice, V.; Despert, G.; Zanna, S.; Josso, P.; Bacos, M.-P.; Marcus, P. XPS study of the initial stages of oxidation of α2-Ti3Al and γ-TiAl intermetallic alloys. Acta Mater. 2007, 55, 3315–3325. [Google Scholar] [CrossRef]
- Shida, Y.; Anada, H. Role of W, Mo, Nb and Si on oxidation of TiAl in air at high temperatures. Mater. Trans. 1994, 35, 623–631. [Google Scholar] [CrossRef]
- Nickel, H.; Zheng, N.X.; Elschner, A.; Quadakkers, W.J. The oxidation behaviour of niobium containing γ-TiAl based intermetallics in air and argon/oxygen. Microchim. Acta 1995, 119, 23–39. [Google Scholar] [CrossRef]
- Shida, Y.; Anada, H. The effect of various ternary additives on the oxidation behavior of TiAl in high-temperature air. Oxid. Met. 1996, 45, 197–219. [Google Scholar] [CrossRef]
- Taniguchi, S.; Shibata, T. Influence of additional elements on the oxidation behaviour of TiAl. Intermetallics 1996, 4, S85–S93. [Google Scholar] [CrossRef]
- Wang, F.H.; Tang, Z.L.; Wu, W.T. Effect of chromium on the oxidation resistance of TiAl intermetallics. Oxid. Met. 1997, 48, 381–390. [Google Scholar] [CrossRef]
- Jiang, H.R.; Wang, Z.L.; Ma, W.S.; Feng, X.R.; Dong, Z.Q.; Zhang, L.; Liu, Y. Effects of Nb and Si on high temperature oxidation of TiAl. Trans. Nonferrous Met. Soc. China 2008, 18, 512–517. [Google Scholar] [CrossRef]
- Vojtěch, D.; Popela, T.; Kubásek, J.; Maixner, J.; Novák, P. Comparison of Nb- and Ta-effectiveness for improvement of the cyclic oxidation resistance of TiAl-based intermetallics. Intermetallics 2011, 19, 493–501. [Google Scholar] [CrossRef]
- Lin, J.P.; Zhao, L.L.; Li, G.Y.; Zhang, L.Q.; Song, X.P.; Ye, F.; Chen, G.L. Effect of Nb on oxidation behavior of high Nb containing TiAl alloys. Intermetallics 2011, 19, 131–136. [Google Scholar] [CrossRef]
- Ping, F.-P.; Hu, Q.-M.; Bakulin, A.V.; Kulkova, S.E.; Yang, R. Alloying effects on properties of Al2O3 and TiO2 in connection with oxidation resistance of TiAl. Intermetallics 2016, 68, 57–62. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.M.; Wang, S.Q.; Ye, H.Q. First-principles study of oxygen atom adsorption on γ-TiAl(111) surface. Acta Metall. Sin. 2006, 42, 897–902. [Google Scholar]
- Li, H.; Wang, S.Q.; Ye, H.Q. Initial oxidation of γ-TiAl(111) surface: Density functional theory study. J. Mater. Sci. Technol. 2009, 25, 569–576. [Google Scholar]
- Liu, S.Y.; Shang, J.X.; Wang, F.H.; Zhang, Y. Ab initio study of surface self-segregation effect on the adsorption of oxygen on the γ-TiAl(111) surface. Phys. Rev. B 2009, 79, 075419. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.Q.; Ye, H.Q. Influence of Nb doping on oxidation resistance of γ-TiAl: A first principles study. Acta Phys. Sin. 2009, 58, S224–S229. [Google Scholar]
- Zhao, C.Y.; Wang, X.; Wang, F.H. First-principles study of Nb doping effect on the diffusion of oxygen atom in γ-TiAl. Adv. Mater. Res. 2011, 304, 148–153. [Google Scholar] [CrossRef]
- Liu, S.Y.; Shang, J.X.; Wang, F.H.; Zhang, Y. Surface segregation of Si and its effect on oxygen adsorption on a γ-TiAl(111) surface from first principles. J. Phys. Condens. Matter 2009, 21, 225005–225007. [Google Scholar] [CrossRef]
- Song, Y.; Dai, J.H. Mechanism of oxygen adsorption on surfaces of γ-TiAl. Surf. Sci. 2012, 606, 852–857. [Google Scholar] [CrossRef]
- Bakulin, A.V.; Kulkova, S.E.; Hu, Q.M.; Yang, R. Adsorption and diffusion of oxygen on (001) and (100) TiAl surface. Comput. Mater. Sci. 2015, 97, 55–63. [Google Scholar] [CrossRef]
- Bakulin, A.V.; Kulkova, S.E.; Hu, Q.M.; Yang, R. Theoretical study of oxygen sorption and diffusion in the volume and on the surface of a γ-TiAl alloy. J. Exp. Theor. Phys. 2015, 120, 257–267. [Google Scholar] [CrossRef]
- Epifano, E.; Hug, G. First-principle study of the solubility and diffusion of oxygen and boron in γ-TiAl. Comput. Mater. Sci. 2020, 174, 107475. [Google Scholar] [CrossRef]
- Connétable, D.; Prillieux, A.; Thenot, C.; Monchoux, J.-P. Theoretical study of oxygen insertion and diffusivity in the γ-TiAl L10 system. J. Phys. Condens. Matter 2020, 32, 175702. [Google Scholar] [CrossRef] [PubMed]
- Bakulin, A.V.; Kulkov, S.S.; Kulkova, S.E. Diffusion properties of oxygen in the γ-TiAl alloy. J. Exp. Theor. Phys. 2020, 130, 579–590. [Google Scholar] [CrossRef]
- Koizumi, Y.; Kishimoto, M.; Minamino, Y.; Nakajima, H. Oxygen diffusion in Ti3Al single crystals. Philos. Mag. 2008, 88, 2991–3010. [Google Scholar] [CrossRef]
- Bakulin, A.V.; Latyshev, A.M.; Kulkova, S.E. Absorption and diffusion of oxygen in the Ti3Al alloy. J. Exp. Theor. Phys. 2017, 125, 138–147. [Google Scholar] [CrossRef]
- Song, Y.; Xing, F.J.; Dai, J.H.; Yang, R. First-principles study of influence of Ti vacancy and Nb dopant on the bonding of TiAl/TiO2 interface. Intermetallics 2014, 49, 1–6. [Google Scholar] [CrossRef]
- Wang, B.D.; Dai, J.H.; Wu, X.; Song, Y.; Yang, R. First-principles study of the bonding characteristics of TiAl(111)/Al2O3(0001) interface. Intermetallics 2015, 60, 58–65. [Google Scholar] [CrossRef]
- Dai, J.H.; Song, Y.; Yang, R. Influence of alloying elements on stability and adhesion ability of TiAl/TiO2 interface by first-principles calculations. Intermetallics 2017, 85, 80–89. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, J. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Beltrán, A.; Andrés, J.; Sambrano, J.R.; Longo, E. Density functional theory study on the structural and electronic properties of low index rutile surfaces for TiO2/SnO2/TiO2 and SnO2/TiO2/SnO2 composite systems. J. Phys. Chem. A 2008, 112, 8943–8952. [Google Scholar] [CrossRef]
- Tanaka, K.; Ichitsubo, T.; Inui, H.; Yamaguchi, M.; Koiwa, M. Single-crystal elastic constants of γ-TiAl. Philos. Mag. Lett. 1996, 73, 71–78. [Google Scholar] [CrossRef]
- Wang, L.; Shang, J.-X.; Wang, F.-H.; Zhang, Y.; Chroneos, A. Unexpected relationship between interlayer distances and surface/cleavage energies in γ-TiAl: Density functional study. J. Phys. Condens. Matter 2011, 23, 265009. [Google Scholar] [CrossRef]
- Wang, J.W.; Gong, H.R. Adsorption and diffusion of hydrogen on Ti, Al, and TiAl surfaces. Int. J. Hydrogen Energy 2014, 39, 6068–6075. [Google Scholar] [CrossRef]
- Ramamoorthy, M.; Vanderbilt, D.; King-Smith, R.D. First-principles calculations of the energetics of stoichiometric TiO2 surfaces. Phys. Rev. B 1994, 49, 16721–16727. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Domain, C.; Roques, J.; Drot, R.; Simoni, E.; Catalette, H. Optimisation of accurate rutile TiO2 (110), (100), (101) and (001) surface models from periodic DFT calculations. Theor. Chem. Acc. 2007, 117, 565–574. [Google Scholar] [CrossRef]
- Moore, D.K.; Cherniak, D.J.; Watson, E.B. Oxygen diffusion in rutile from 750 to 1000 °C and 0.1 to 1000 MPa. Am. Mineral. 1998, 83, 700–711. [Google Scholar] [CrossRef]
- Herzig, C.; Przeorski, T.; Mishin, Y. Self-diffusion in γ-TiAl: An experimental study and atomistic calculations. Intermetallics 1999, 7, 389–404. [Google Scholar] [CrossRef]
- Zhang, W.; Smith, J.R.; Evans, A.G. The connection between ab initio calculations and interface adhesion measurements on metal/oxide systems: Ni/Al2O3 and Cu/Al2O3. Acta Mater. 2002, 50, 3803–3816. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, W.; Jiang, W. Ab initio study of Ag/Al2O3 and Au/Al2O3 interfaces. Phys. Rev. B 2005, 72, 115423. [Google Scholar] [CrossRef]
- Melnikov, V.V.; Eremeev, S.V.; Kulkova, S.E. Adhesion of niobium films to differently oriented α-Al2O3 surfaces. Tech. Phys. 2011, 56, 1494–1500. [Google Scholar] [CrossRef]
- Du, J.L.; Fang, Y.; Fu, E.G.; Ding, X.; Yu, K.Y.; Wang, Y.G.; Wang, Y.Q.; Baldwin, J.K.; Wang, P.P.; Bai, Q. What determines the interfacial configuration of Nb/Al2O3 and Nb/MgO interface. Sci. Rep. 2006, 6, 33931. [Google Scholar] [CrossRef]
- Mei, Z.-G.; Bhattacharya, S.; Yacout, A.M. Adhesion of ZrN and Al2O3 coatings on U metal from first-principles. Appl. Surf. Sci. 2019, 473, 121–126. [Google Scholar] [CrossRef]
- Manz, T.A.; Limas, N.G. Introducing DDEC6 atomic population analysis: Part 1. Charge partitioning theory and methodology. RSC Adv. 2016, 6, 47771–47801. [Google Scholar] [CrossRef]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 atomic population analysis: Part 2. Computed results for a wide range of periodic and nonperiodic materials. RSC Adv. 2016, 6, 45727–45747. [Google Scholar] [CrossRef]
- Manz, T.A. Introducing DDEC6 atomic population analysis: Part 3. Comprehensive method to compute bond orders. RSC Adv. 2017, 7, 45552–45581. [Google Scholar] [CrossRef]
- Griffith, A.A. The phenomena of rupture and ow in solids. Trans. R. Soc. Lond. 1921, A221, 163–198. [Google Scholar] [CrossRef]
- Zehnder, A. Griffith theory of fracture. In Encyclopedia of Tribology; Wang, Q.J., Chung, Y.-W., Eds.; Springer: New York, NY, USA, 2013; pp. 1570–1573. [Google Scholar] [CrossRef]
- Bakulin, A.V.; Kulkov, S.S.; Kulkova, S.E. Adhesive properties of the TiAl/Al2O3 interface. Russ. Phys. J. 2020, in press. [Google Scholar] [CrossRef]
- Bakulin, A.V.; Kulkov, S.S.; Kulkova, S.E. Adhesion properties of clean and doped Ti3Al/Al2O3 interface. Appl. Surf. Sci. 2020, 536, 147639. [Google Scholar] [CrossRef]
- Bakulin, A.V.; Kulkova, S.E. Adhesion at the Ta(Mo)/NiTi Interface. Tech. Phys. Lett. 2019, 45, 620–624. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | TiAl | Compound | TiO2 | |||||
|---|---|---|---|---|---|---|---|---|
| Surface | (100) | (111) | (011) | Surface | (110)O | (100)O | (001) | (011)O |
| Present result PAW-PBE | 1.69 | 1.71 | 2.10 | Present result PAW-PBE | 0.46 | 0.62 | 1.20 | 1.02 |
| [40] PAW-GGA | 1.62 | 1.69 | [28] PAW-GGA | 0.46 | 0.56 | 1.44 | 1.05 | |
| [41] PAW-LDA | 1.91 | [42] PAW-GGA | 0.50 | 0.69 | 1.25 | 1.03 | ||
| [43] US PP LDA | 0.89 | 1.12 | 1.40 | 1.65 | ||||
| Alloy Termination | Configuration | Wsep | d |
|---|---|---|---|
| Al | hollow | 1.27 | 0.75 |
| top | 0.74 | 1.38 | |
| [28] | 0.58–1.64 | ||
| Ti | hollow | 2.44 | 0.62 |
| top | 1.97 | 0.50 | |
| [30] | 4.97 |
| Atom | Q | d | θ | Atom | Q | d | θ |
|---|---|---|---|---|---|---|---|
| TiAl(110)Al/TiO2(110)O | TiAl(110)Ti/TiO2(110)O | ||||||
| Ti1I+1 O1I | +1.99 –0.93 | 2.20 | 0.183 | Ti1I+1 O1I | +2.00 –1.00 | 2.07 | 0.253 |
| Ti2I+1 O2I | +2.01 –0.92 | 2.07 | 0.255 | Ti2I+1 O2I | +1.89 –0.83 | 2.23 | 0.183 |
| O1I Al1I | –0.93 +0.69 | 1.89, 1.78 [28] | 0.481 | O1I Ti1I | –1.00 +0.82 | 1.98, 2.22–2.31 [30] | 0.483 |
| O2I TiI–1 | –0.92 +0.42 | 1.92, 2.08 [28] | 0.594 | O2I AlI–1 | –0.83 –0.02 | 2.07, 1.90 [30] | 0.403 |
| OI+1 Al2I | –1.00 +0.19 | 1.90, 1.85 [28] | 0.559 | OI+1 Ti2I | –1.14 +0.51 | 2.04, 1.92 [30] | 0.505 |
| TiAl(110)Al surface 1 | TiAl(110)Ti surface | ||||||
| AlS TiS–1 | –0.24 +0.29 | 2.90 | 0.277 | TiS AlS–1 | +0.17 –0.30 | 2.84 | 0.302 |
| TiO2(110)O surface | |||||||
| OS TiS+1 | –0.88 +2.13 | 1.85 | 0.406 | ||||
| OS+1 TiS+1 | –1.13 +2.13 | 1.95–2.03 | 0.279–0.340 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakulin, A.V.; Kulkov, S.S.; Kulkova, S.E.; Hocker, S.; Schmauder, S. First Principles Study of Bonding Mechanisms at the TiAl/TiO2 Interface. Metals 2020, 10, 1298. https://doi.org/10.3390/met10101298
Bakulin AV, Kulkov SS, Kulkova SE, Hocker S, Schmauder S. First Principles Study of Bonding Mechanisms at the TiAl/TiO2 Interface. Metals. 2020; 10(10):1298. https://doi.org/10.3390/met10101298
Chicago/Turabian StyleBakulin, Alexander V., Sergey S. Kulkov, Svetlana E. Kulkova, Stephen Hocker, and Siegfried Schmauder. 2020. "First Principles Study of Bonding Mechanisms at the TiAl/TiO2 Interface" Metals 10, no. 10: 1298. https://doi.org/10.3390/met10101298
APA StyleBakulin, A. V., Kulkov, S. S., Kulkova, S. E., Hocker, S., & Schmauder, S. (2020). First Principles Study of Bonding Mechanisms at the TiAl/TiO2 Interface. Metals, 10(10), 1298. https://doi.org/10.3390/met10101298