
Antibiotic Therapy and Athletes: Is the Mitochondrial Dysfunction the Real Achilles’ Heel?
Abstract
:1. Introduction
2. Antibiotics: Classification, Mechanisms of Action, and Adverse Effects
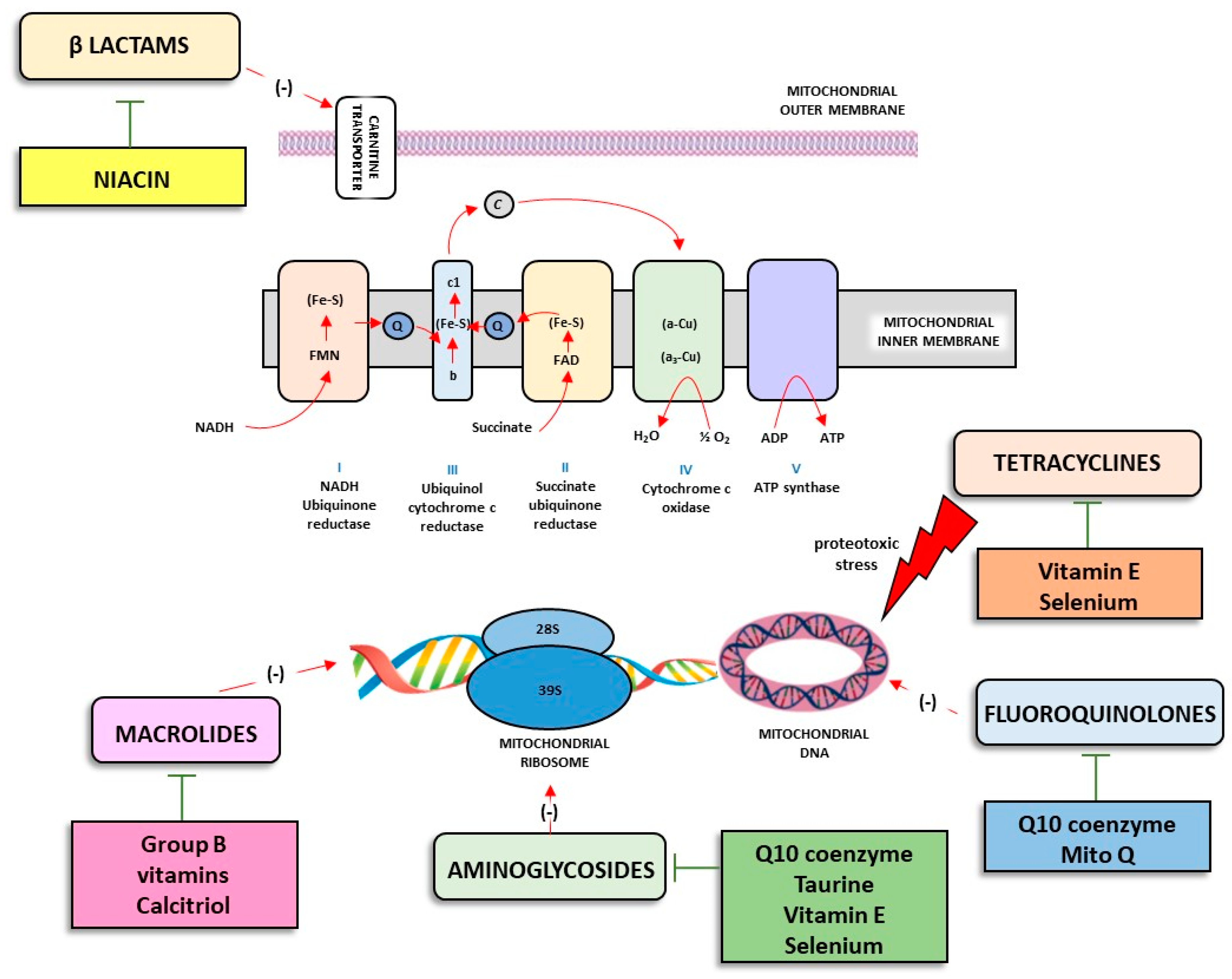
3. Antibiotics and Altered Mitochondria Physiology in Skeletal Muscle
4. Antibiotics and Tendinopathy: The Real Achilles Heel?
5. Therapeutic Strategies Aimed at Counteracting Tendon and Muscle Alterations Associated with Antibiotic Use
6. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Fayock, K.; Voltz, M.; Sandella, B.; Close, J.; Lunser, M.; Okon, J. Antibiotic Precautions in Athletes. Sports Health 2014, 6, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Aavikko, A.; Helenius, I.; Vasankari, T.; Alaranta, A. Physician-Prescribed Medication Use by the Finnish Paralympic and Olympic Athletes. Clin. J. Sport Med. 2013, 23, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Tiernan, C.; Comyns, T.; Lyons, M.; Nevill, A.M.; Warrington, G. The Association Between Training Load Indices and Injuries in Elite Soccer Players. J. Strength Cond. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.; Cook, J. Fluoroquinolones and Tendinopathy: A Guide for Athletes and Sports Clinicians and a Systematic Review of the Literature. J. Athl. Train. 2014, 49, 422–427. [Google Scholar] [CrossRef]
- Pancu, D.F.; Scurtu, A.; Macasoi, I.G.; Marti, D.; Mioc, M.; Soica, C.; Coriovac, D.; Horhat, D.; Poenaru, M.; Dehelean, C. Antibiotics: Conventional Therapy and Natural Compounds with Antibacterial Activity-A Pharma-co-Toxicological Screening. Antibiotics 2021, 10, 401. [Google Scholar] [CrossRef]
- Hooper, D.C. Mechanisms of Action of Antimicrobials: Focus on Fluoroquinolones. Clin. Infect. Dis. 2001, 32 (Suppl. S1), S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal Muscle Fatigue: Cellular Mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [CrossRef]
- Boguszewska, K.; Szewczuk, M.; Kaźmierczak-Barańska, J.; Karwowski, B.T. The Similarities between Human Mitochondria and Bacteria in the Context of Structure, Genome, and Base Excision Repair System. Molecules 2020, 25, 2857. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal Antibiotics Induce Mitochondrial Dysfunction and Oxidative Damage in Mammalian Cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Asai, Y.; Kagi, T.; Noguchi, T.; Yamada, M.; Hirata, Y.; Matsuzawa, A. TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms. Int. J. Mol. Sci. 2020, 21, 9497. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Galluccio, M.; Scumaci, D.; Giangregorio, N.; Tonazzi, A.; Palmieri, F.; Indiveri, C. Interaction of beta-lactam antibiotics with the mitochondrial carnitine/acylcarnitine transporter. Chem. Biol. Interact. 2008, 173, 187–194. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Indiveri, C. Mitochondrial Carnitine/Acylcarnitine Translocase: Insights in Structure/Function Relationships. Basis for drug therapy and side effects prediction. Mini Rev. Med. Chem. 2015, 5, 396–405. [Google Scholar] [CrossRef]
- Ojano-Dirain, C.P.; Antonelli, P.J. Prevention of gentamicin-induced apoptosis with the mitochondria-targeted antioxidant mitoquinone. Laryngoscope 2012, 122, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M.; Simmons, F.; Humes, H.D. Alterations of mitochondrial respiration induced by aminoglycoside antibiotics. Res. Commun. Chem. Pathol. Pharmacol. 1980, 27, 521–531. [Google Scholar]
- Guerrieri, F.; Micelli, S.; Massagli, C.; Gallucci, E.; Papa, S. Interaction of the aminoglycoside antibiotic dihydrostreptomycin with the H+-ATPase of mitochondria. Biochem. Pharmacol. 1984, 33, 2505–2510. [Google Scholar] [CrossRef]
- O’Reilly, M.; Young, L.; Kirkwood, N.K.; Richardson, G.P.; Kros, C.J.; Moore, A.L. Gentamicin Affects the Bioenergetics of Isolated Mitochondria and Collapses the Mitochondrial Membrane Potential in Cochlear Sensory Hair Cells. Front. Cell. Neurosci. 2019, 13, 416. [Google Scholar] [CrossRef]
- Dupont-Versteegden, E.E. Apoptosis in skeletal muscle and its relevance to atrophy. World J. Gastroenterol. 2006, 12, 7463–7466. [Google Scholar] [CrossRef] [PubMed]
- Ojano-Dirain, C.P.; Antonelli, P.J.; Le Prell, C.G. Mitochondria-Targeted Antioxidant MitoQ Reduces Gentamicin-Induced Ototoxicity. Otol. Neurotol. 2014, 35, 533–539. [Google Scholar] [CrossRef]
- Müller-Höcker, J. Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: An age-related alteration. J. Neurol. Sci. 1990, 100, 14–21. [Google Scholar] [CrossRef]
- Barron, M.J.; Chinnery, P.; Howel, D.; Blakely, E.; Schaefer, A.; Taylor, R.; Turnbull, D. Cytochrome c oxidase deficient muscle fibres: Substantial variation in their proportions within skeletal muscles from patients with mitochondrial myopathy. Neuromuscul. Disord. 2005, 15, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Kadenbach, B.; Droste, M.; Old, S.; Turnbull, D. Immunocytochemical studies of cytochrome oxidase subunits in skeletal muscle of patients with partial cytochrome oxidase deficiencies. J. Neurol. Sci. 1988, 87, 75–90. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Kiriaev, L.; Perry, B.D.; Mahns, D.A.; Shortland, P.J.; Redwan, A.; Morley, J.W.; Head, S.I. Minocycline Treatment Reduces Mass and Force Output from Fast-Twitch Mouse Muscles and Inhibits Myosin Production in C2C12 Myotubes. Front. Physiol. 2021, 12, 696039. [Google Scholar] [CrossRef] [PubMed]
- Shefer, G.; Carmeli, E.; Rauner, G.; Yablonka-Reuveni, Z.; Benayahu, D. Exercise running and tetracycline as means to enhance skeletal muscle stem cell performance after external fixation. J. Cell. Physiol. 2008, 215, 265–275. [Google Scholar] [CrossRef]
- Obradovic, H.; Krstić, J.; Kukolj, T.; Trivanović, D.; Đorđević, I.O.; Mojsilović, S.; Jauković, A.; Jovčić, G.; Bugarski, D.; Santibañez, J.F. Doxycycline Inhibits IL-17-Stimulated MMP-9 Expression by Downregulating ERK1/2 Activation: Implications in Myogenic Differentiation. Mediators Inflamm. 2016, 2016, 2939658. [Google Scholar] [CrossRef]
- Tribuiani, N.; de Souza, J.; de Queiroz Junior, M.A.; Baldo, D.A.; de Campos Orsi, V.; Oshima-Franco, Y. Effects of Doxycycline On Mice Neuromuscular Junction, In Situ. Curr. Mol. Med. 2021, 22, 349–353. [Google Scholar] [CrossRef]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Bio-medical Research. Cell. Rep. 2015, 10, 1681–1691. [Google Scholar] [CrossRef]
- Hall, M.M.; Finnoff, J.T.; Smith, J. Musculoskeletal Complications of Fluoroquinolones: Guidelines and Precautions for Usage in the Athletic Population. PM&R 2011, 3, 132–142. [Google Scholar] [CrossRef]
- Guis, S.; Jouglard, J.; Kozak-Ribbens, G.; Figarella-Branger, D.; Vanuxem, D.; Pellissier, J.F.; Cozzone, P.J. Malignant hyperthermia susceptibility revealed by myalgia and rhabdomyolysis during fluoroquinolone treatment. J. Rheumatol. 2001, 28, 1405–1406. [Google Scholar] [PubMed]
- Hangas, A.; Aasumets, K.; Kekäläinen, N.J.; Paloheinä, M.; Pohjoismäki, J.L.; Gerhold, J.M.; Goffart, S. Ciprofloxacin impairs mitochondrial DNA replication initiation through inhibition of Topoisomerase 2. Nucleic Acids Res. 2018, 46, 9625–9636. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef]
- Gupta, A.; Ökesli-Armlovich, A.; Morgens, D.; Bassik, M.C.; Khosla, C. A genome-wide analysis of targets of macrolide antibiotics in mammalian cells. J. Biol. Chem. 2020, 295, 2057–2067. [Google Scholar] [CrossRef]
- Gindre, H.; Féasson, L.; Auboyer, C.; Cathébras, P. Severe rhabdomyolysis associated with a primary cytomegalovirus infection in an immunocompetent patient. BMJ Case Rep. 2013, 2013, bcr2012008140. [Google Scholar] [CrossRef]
- Michalak, K.; Sobolewska-Włodarczyk, A.; Włodarczyk, M.; Sobolewska, J.; Wozniak, P.; Sobolewski, B. Treatment of the Fluoroquinolone-Associated Disability: The Pathobiochemical Implications. Oxid. Med. Cell. Longev. 2017, 2017, 8023935. [Google Scholar] [CrossRef] [PubMed]
- Childs, S.G. Pathogenesis of Tendon Rupture Secondary to Fluoroquinolone Therapy. Orthop. Nurs. 2007, 26, 175–182, quiz 183-4. [Google Scholar] [CrossRef]
- Stahlmann, R.; Lode, H. Toxicity of quinolones. Drugs 1999, 58 (Suppl. S2), 37–42. [Google Scholar] [CrossRef] [PubMed]
- Reuveni, D.; Halperin, D.; Shalit, I.; Priel, E.; Fabian, I. Quinolones as enhancers of camptothecin-induced cytotoxic and anti-topoisomerase I effects. Biochem. Pharmacol. 2008, 75, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Reuveni, D.; Halperin, D.; Fabian, I.; Tsarfaty, G.; Askenasy, N.; Shalit, I. Moxifloxacin increases anti-tumor and anti-angiogenic activity of irinotecan in human xenograft tumors. Biochem. Pharmacol. 2010, 79, 1100–1107. [Google Scholar] [CrossRef]
- Kim, K.-O.; Sim, J.A.; Choi, J.U.; Lee, B.K.; Park, H.G. The effect of interleukin-8 in the early stage after anterior cruciate ligament reconstruction with remnant preservation. Knee Surg. Relat. Res. 2020, 32, 5. [Google Scholar] [CrossRef] [PubMed]
- Screen, H.R.C.; Berk, D.E.; Kadler, K.E.; Ramirez, F.; Young, M.F. Tendon Functional Extracellular Matrix. J. Orthop. Res. 2015, 33, 793–799. [Google Scholar] [CrossRef]
- Sendzik, J.; Shakibaei, M.; Schäfer-Korting, M.; Stahlmann, R. Fluoroquinolones cause changes in extracellular matrix, signalling proteins, metalloproteinases and caspase-3 in cultured human tendon cells. Toxicology 2005, 212, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Shakibaei, M.; De Souza, P.; Van Sickle, D.; Stahlmann, R. Biochemical changes in Achilles tendon from juvenile dogs after treatment with ciprofloxacin or feeding a magnesium-deficient diet. Arch. Toxicol. 2001, 75, 369–374. [Google Scholar] [CrossRef]
- Badal, S.; Her, Y.F.; Maher, L.J. Nonantibiotic Effects of Fluoroquinolones in Mammalian Cells. J. Biol. Chem. 2015, 290, 22287–22297. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J., 3rd; Attia, E.; Wickiewicz, T.L.; Hannafin, J.A. The effect of ciprofloxacin on tendon, paratenon, and capsular fibroblast metabolism. Am. J. Sports Med. 2000, 28, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.; Pettinari, L.; Martinelli, C.; Colombo, G.; Portinaro, N.; Dalle-Donne, I.; D’Agostino, M.C.; Gagliano, N. New insights in extracellular matrix remodeling and collagen turnover related pathways in cultured human tenocytes after ciprofloxacin administration. Muscles Ligaments Tendons J. 2013, 3, 122–131. [Google Scholar]
- Tsai, W.-C.; Hsu, C.-C.; Chen, C.P.; Chang, H.-N.; Wong, A.M.; Lin, M.-S.; Pang, J.-H.S. Ciprofloxacin up-regulates tendon cells to express matrix metalloproteinase-2 with degradation of type I collagen. J. Orthop. Res. 2011, 29, 67–73. [Google Scholar] [CrossRef]
- Corps, A.N.; Harrall, R.L.; Curry, V.A.; Fenwick, S.A.; Hazleman, B.L.; Riley, G.P. Ciprofloxacin enhances the stimulation of matrix metalloproteinase 3 expression by interleukin-1beta in human tendon-derived cells. A potential mechanism of fluoroquinolone-induced tendinopathy. Arthritis Rheum. 2002, 46, 3034–3040. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular Strategies for Targeting Antioxidants to Mitochondria: Therapeutic Implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Sugahara, K.; Hirose, Y.; Mikuriya, T.; Hashimoto, M.; Kanagawa, E.; Hara, H.; Shimogori, H.; Yamashita, H. Coenzyme Q10 Protects Hair Cells against Aminoglycoside. PLoS ONE 2014, 9, e108280. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Chi, F.-L.; Gao, W.-Y. Taurine attenuates aminoglycoside ototoxicity by inhibiting inducible nitric oxide synthase expression in the cochlea. NeuroReport 2008, 19, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Kharkheli, E.; Kevanishvili, Z.; Maglakelidze, T.; Davitashvili, O.; Schacht, J. Does vitamin E prevent gentamicin-induced ototoxicity? Georgian Med. News 2007, 146, 14–17. [Google Scholar]
- Le Prell, C.G.; Ojano-Dirain, C.; Rudnick, E.W.; Nelson, M.A.; DeRemer, S.J.; Prieskorn, D.M.; Miller, J.M. Assessment of Nutrient Supplement to Reduce Gentamicin-Induced Ototoxicity. J. Assoc. Res. Otolaryngol. 2014, 15, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Ngaha, E.O.; Ogunleye, I.O. Studies on gentamicin-induced labilization of rat kidney lysosomes in vitro. Possible protection by selenium. Biochem. Pharmacol. 1983, 32, 2659–2664. [Google Scholar] [CrossRef]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O’Brien, P.J. Mitochondrial function and toxicity: Role of B vitamins on the one-carbon transfer pathways. Chem. Biol. Interact. 2006, 163, 113–132. [Google Scholar] [CrossRef]
- Lowes, D.A.; Wallace, C.; Murphy, M.P.; Webster, N.R.; Galley, H.F. The mitochondria targeted antioxidant MitoQ protects against fluoroquinolone-induced oxidative stress and mitochondrial membrane damage in human Achilles tendon cells. Free Radic. Res. 2009, 43, 323–328. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Drugs | Mechanisms of Action | Resistance Mechanisms | Adverse Events | Ref. |
|---|---|---|---|---|
| β lactams | Bactericidal. Inhibition of bacteria cell wall synthesis interacting with penicillin-binding protein (PBP). |
| Diarrhea, nausea, and vomiting Abdominal pain Allergic reactions Hypersensitivity | [1] |
| Aminoglycosides | Bactericidal. Binding with 30S ribosomal subunit. Reduced translation. Inhibition of protein synthesis. |
| Headache Paresthesia Fever Superinfections Vertigo Skin rash Dizziness | [2] |
| Tetracyclines | Bacteriostatic. Binding with 30S ribosomal subunit. Reduced translation. Inhibition of protein synthesis. |
| Discoloration of teeth Enamel hypoplasia Diarrhea Nausea Photosensitivity Stomach upset Loss of appetite White patches or sores inside mouth or on lips Swollen tongue | [3] |
| Fluoroquinolones | Bactericidal. Targets of GyrA subunit of DNA gyrase and topoisomerase IV. Inhibition of DNA synthesis |
| Digestive disorders Hyperglycemia Hypoglycemia QTc prolongation and cardiac arrhythmia Retinal detachment Tendinopathy Tendon rupture Peripheral neuropathy Aortic aneurysm | [4] |
| Macrolides | Bacteriostatic. Binding with 50S ribosomal subunit. Inhibition of protein synthesis. |
| Allergic reactions Cholestatic hepatitis Cardiac arrhythmias Transient auditory impairment | [5] |
| Carbapenems | Bactericidal. Atypical β-lactam antibiotics with broad-spectrum high antibacterial activity. | extended spectrum β-lactamases | Nausea and vomiting Seizures Patients with allergies to other β-lactams may experience hypersensitivity reactions | [6] |
| Trimethoprim | Bacteriostatic. Inhibition of folate synthesis. Dihydrofolate reductase (DHFR) inhibition. |
| Itching and rash Diarrhea Nausea Vomiting Stomach upset Loss of appetite Changes in taste Headache Skin sensitivity to sunlight Swollen tongue Fever | [7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puccini, V. Antibiotic Therapy and Athletes: Is the Mitochondrial Dysfunction the Real Achilles’ Heel? Sports 2022, 10, 131. https://doi.org/10.3390/sports10090131
Puccini V. Antibiotic Therapy and Athletes: Is the Mitochondrial Dysfunction the Real Achilles’ Heel? Sports. 2022; 10(9):131. https://doi.org/10.3390/sports10090131
Chicago/Turabian StylePuccini, Valentina. 2022. "Antibiotic Therapy and Athletes: Is the Mitochondrial Dysfunction the Real Achilles’ Heel?" Sports 10, no. 9: 131. https://doi.org/10.3390/sports10090131
APA StylePuccini, V. (2022). Antibiotic Therapy and Athletes: Is the Mitochondrial Dysfunction the Real Achilles’ Heel? Sports, 10(9), 131. https://doi.org/10.3390/sports10090131