

Exploring Epigenetic Ageing Using Direct Methylome Sequencing

 ,
,  and
and
Abstract
1. Introduction
2. Results
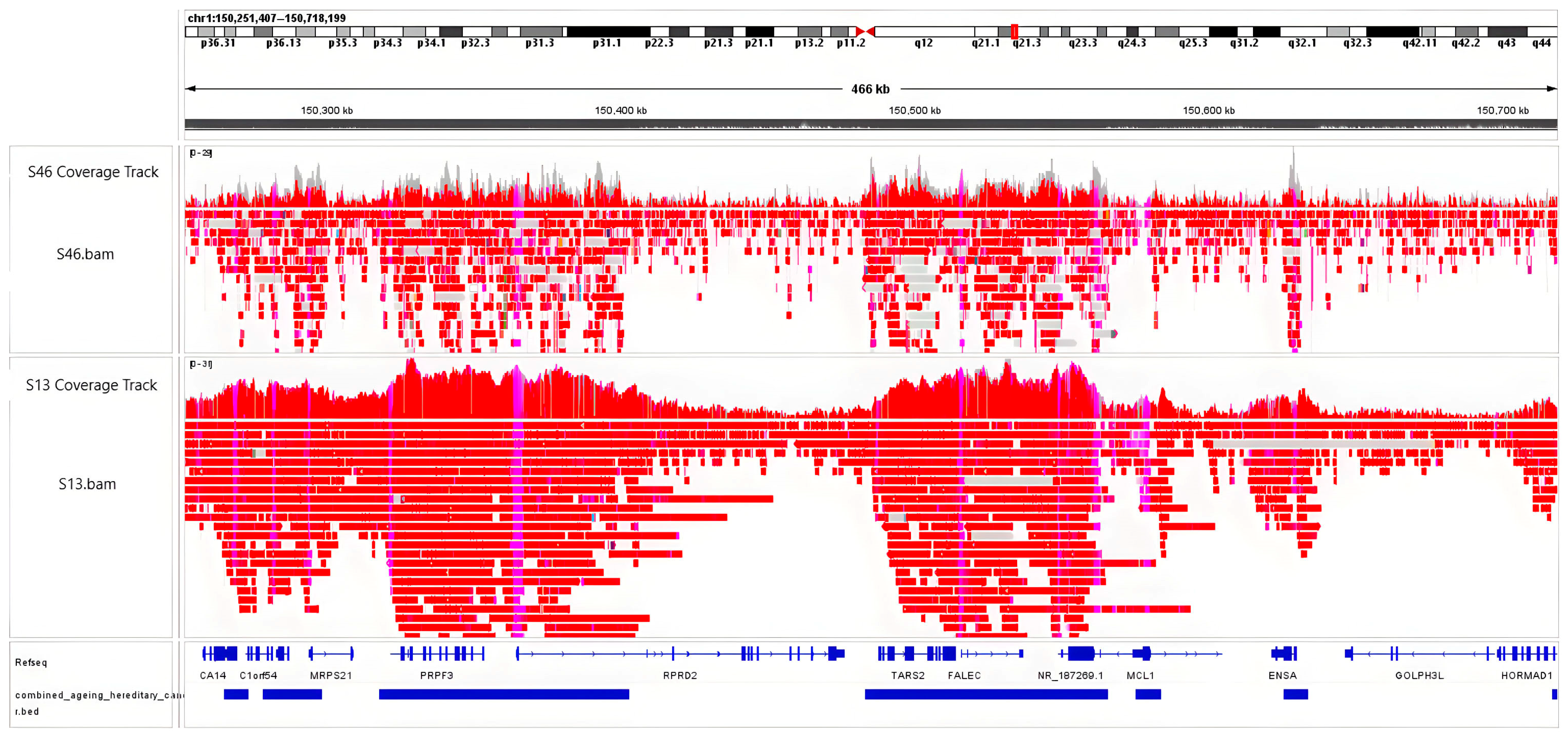
2.1. Enrichement of Genomic Target Regions with Adaptive Sampling
2.2. The DNA Methylation Landscape Across the Lifespan
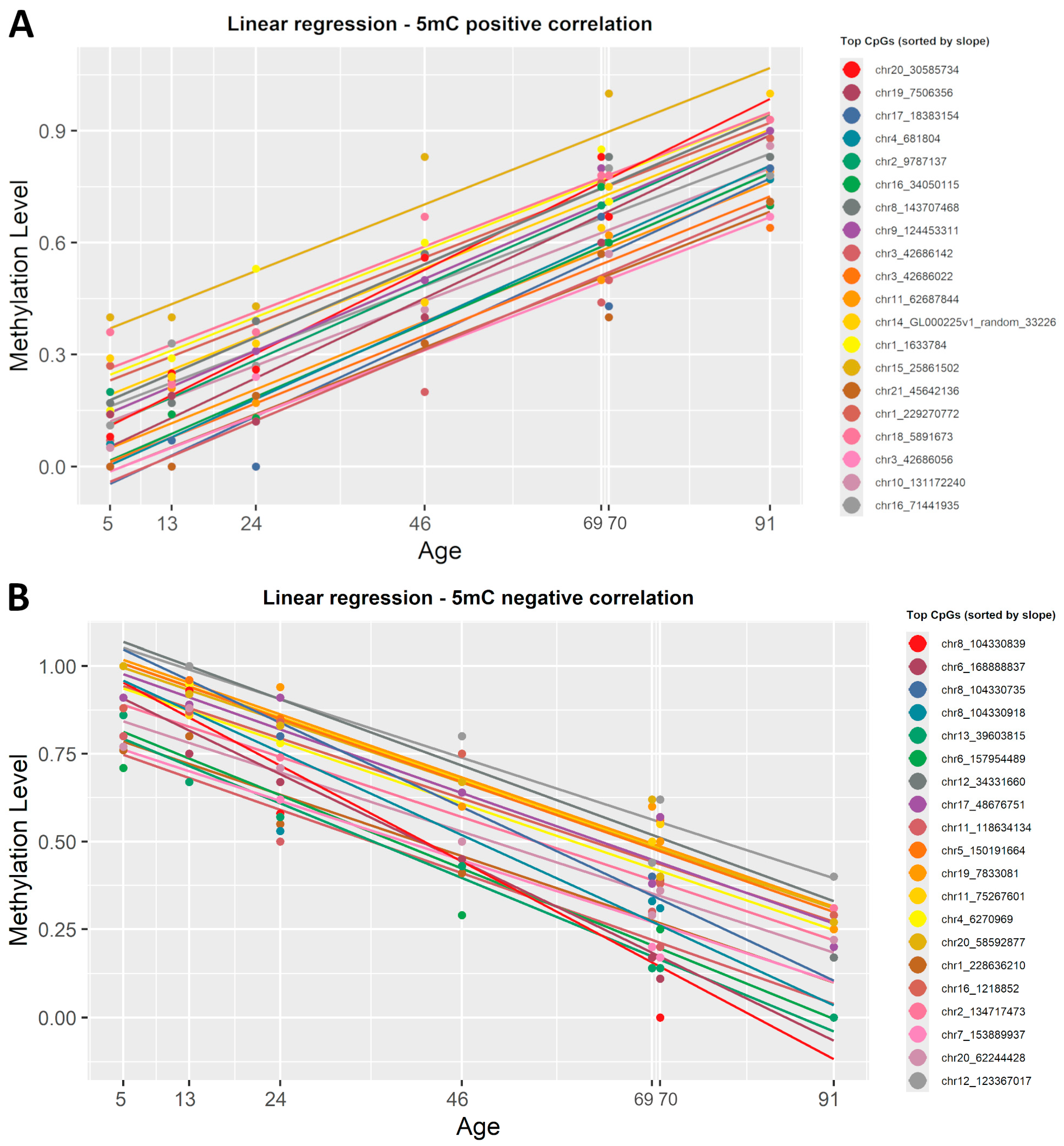
2.3. Detecting Age-Related DNA Methylation Changes with a Simple Linear Regression
2.4. Detecting Age-Related DNA Methylation Changes with ElasticNet Regression
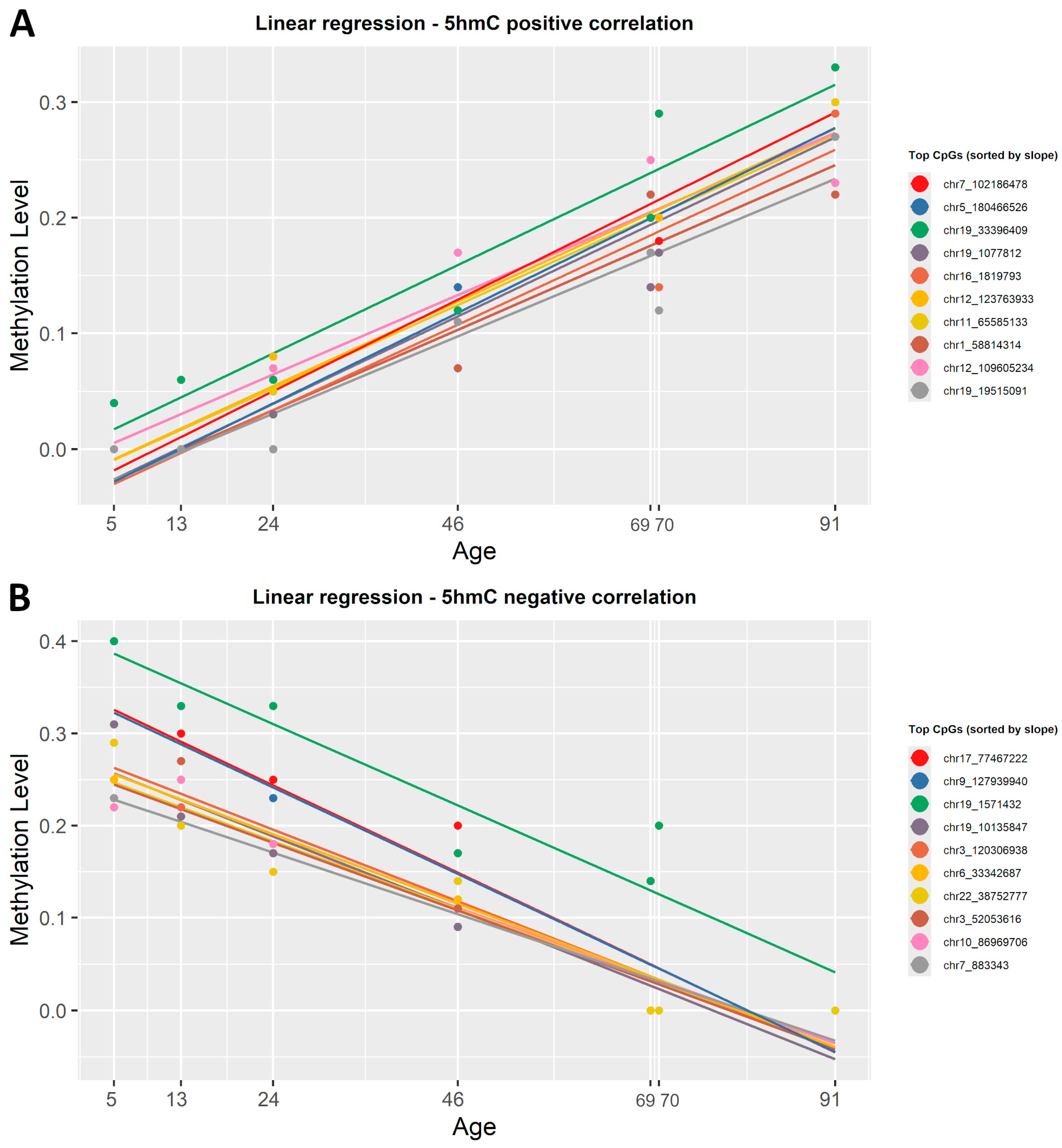
2.5. Detecting Age-Related Changes in the Level of 5hmC with Linear Regression
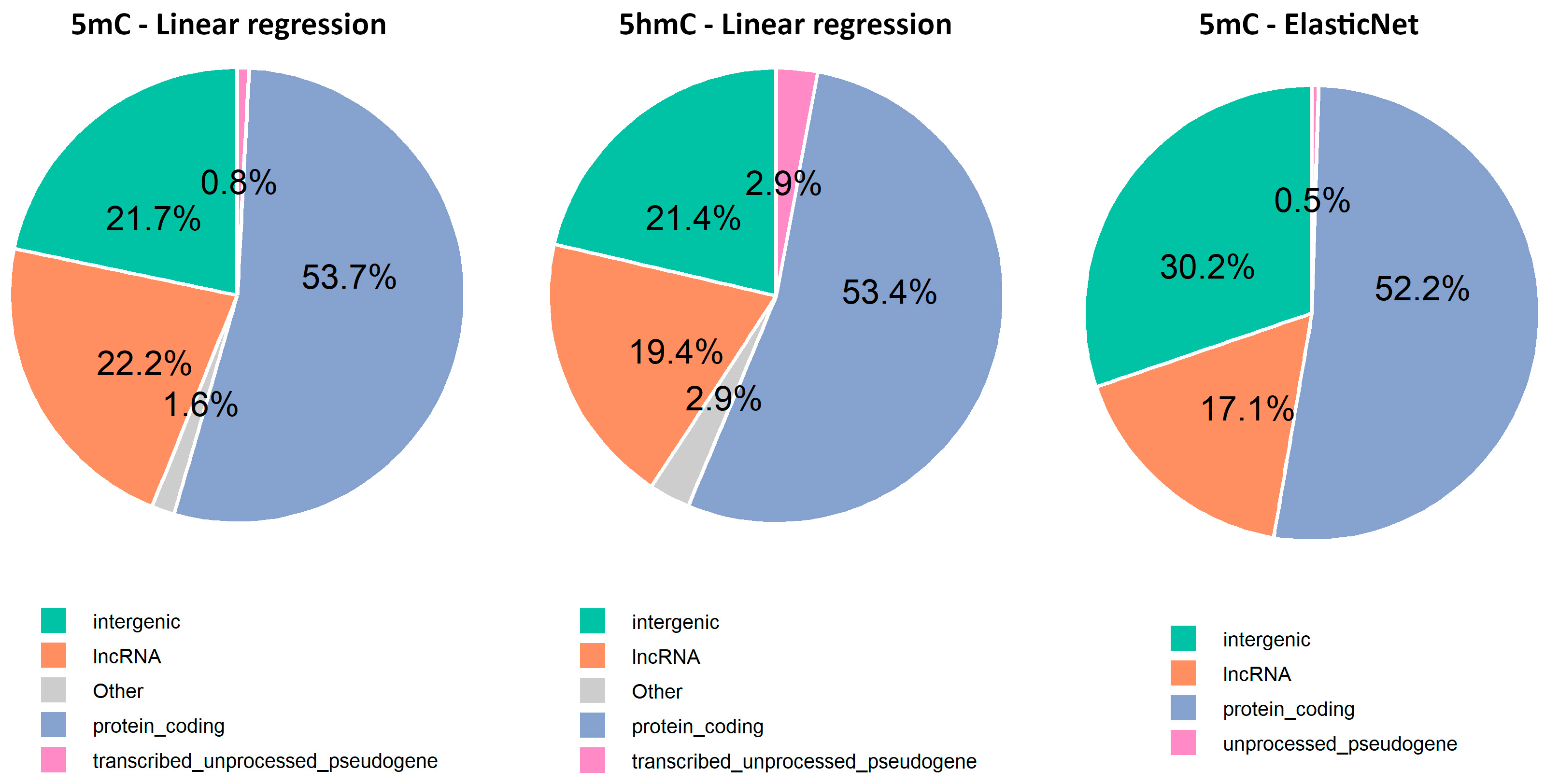
2.6. Exploring New Methylation Markers and Their Genomic Locations
3. Discussion
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. DNA Purification and Fragmentation
4.3. Adaptive Sampling Target Panel Assembly
4.4. DNA Library Preparation and Adaptive Sampling Sequencing
4.5. Sequencing Data Processing and DNA Methylation Analysis
4.6. Regression Models
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Browder, K.C.; Reddy, P.; Yamamoto, M.; Haghani, A.; Guillen, I.G.; Sahu, S.; Wang, C.; Luque, Y.; Prieto, J.; Shi, L.; et al. In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat. Aging 2022, 2, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Rollandi, G.A.; Chiesa, A.; Sacchi, N.; Castagnetta, M.; Puntoni, M.; Amaro, A.; Banelli, B.; Pfeffer, U. Biological Age Versus Chronological Age in the Prevention of Age Associated Diseases. OBM Geriatr. 2019, 3, 051. [Google Scholar] [CrossRef]
- Sarink, D.; Nedkoff, L.; Briffa, T.; Shaw, J.E.; Magliano, D.J.; Stevenson, C.; Mannan, H.; Knuiman, M.; Hung, J.; Hankey, G.J.; et al. Trends in age- and sex-specific prevalence and incidence of cardiovascular disease in Western Australia. Eur. J. Prev. Cardiol. 2018, 25, 1280–1290. [Google Scholar] [CrossRef]
- Karg, M.M.; Lu, Y.R.; Refaian, N.; Cameron, J.; Hoffmann, E.; Hoppe, C.; Shirahama, S.; Shah, M.; Krasniqi, D.; Krishnan, A.; et al. Sustained Vision Recovery by OSK Gene Therapy in a Mouse Model of Glaucoma. Cell. Reprogram. 2023, 25, 288–299. [Google Scholar] [CrossRef]
- Macdonald-Dunlop, E.; Taba, N.; Klarić, L.; Frkatović, A.; Walker, R.; Hayward, C.; Esko, T.; Haley, C.; Fischer, K.; Wilson, J.F.; et al. A catalogue of omics biological ageing clocks reveals substantial commonality and associations with disease risk. Aging 2022, 14, 623–659. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef]
- Lister-Shimauchi, E.H.; McCarthy, B.; Lippincott, M.; Ahmed, S. Genetic and Epigenetic Inheritance at Telomeres. Epigenomes 2022, 6, 9. [Google Scholar] [CrossRef]
- Peng, C.; Wang, X.; Chen, J.; Jiao, R.; Wang, L.; Li, Y.M.; Zuo, Y.; Liu, Y.; Lei, L.; Ma, K.Y.; et al. Biology of ageing and role of dietary antioxidants. BioMed Res. Int. 2014, 2014, 831841. [Google Scholar] [CrossRef]
- Xia, X.; Chen, W.; McDermott, J.; Han, J.D.J. Molecular and phenotypic biomarkers of aging. F1000Research 2017, 6, 860. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Page, A.L.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and inflamm-aging as two sides of the same coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, X.; Luo, J.; Bao, T.; Wang, S.; Wu, X. Molecular mechanisms of aging and anti-aging strategies. Cell Commun. Signal. 2024, 22, 285. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.V.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Tan, Q.; Alo, H.; Nygaard, M.; Sørensen, M.; Saleh, A.; Mengel-From, J.; Christensen, K. Age-Dependent DNA Methylation Variability on the X-Chromosome in Male and Female Twins. Epigenomes 2024, 8, 43. [Google Scholar] [CrossRef]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733. [Google Scholar] [CrossRef]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 2019, 18, e12877. [Google Scholar] [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Chondronasiou, D.; Gill, D.; Mosteiro, L.; Urdinguio, R.G.; Berenguer-Llergo, A.; Aguilera, M.; Durand, S.; Aprahamian, F.; Nirmalathasan, N.; Abad, M.; et al. Multi-omic rejuvenation of naturally aged tissues by a single cycle of transient reprogramming. Aging Cell 2022, 21, e13578. [Google Scholar] [CrossRef]
- Alle, Q.; Le Borgne, E.; Bensadoun, P.; Lemey, C.; Béchir, N.; Gabanou, M.; Estermann, F.; Bertrand-Gaday, C.; Pessemesse, L.; Toupet, K.; et al. A single short reprogramming early in life initiates and propagates an epigenetically related mechanism improving fitness and promoting an increased healthy lifespan. Aging Cell 2022, 21, e13714. [Google Scholar] [CrossRef]
- Macip, C.C.; Hasan, R.; Hoznek, V.; Kim, J.; Lu, Y.R.; Metzger, L.E.; Sethna, S.; Davidsohn, N. Gene Therapy-Mediated Partial Reprogramming Extends Lifespan and Reverses Age-Related Changes in Aged Mice. Cell. Reprogram. 2024, 26, 24–32. [Google Scholar] [CrossRef]
- Seale, K.; Horvath, S.; Teschendorff, A.; Eynon, N.; Voisin, S. Making sense of the ageing methylome. Nat. Rev. Genet. 2022, 23, 585–605. [Google Scholar] [CrossRef]
- Ahsan, M.U.; Gouru, A.; Chan, J.; Zhou, W.; Wang, K. A signal processing and deep learning framework for methylation detection using Oxford Nanopore sequencing. Nat. Commun. 2024, 15, 1448. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar]
- Bernstein, C. Epigenetic field defects in progression to cancer. World J. Gastrointest. Oncol. 2013, 5, 43. [Google Scholar] [CrossRef]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 54. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, Z.; Wang, J.; Chiu, B.C.; Hou, L.; Zhang, W. Application of the High-Throughput TAB-Array for the Discovery of Novel 5-Hydroxymethylcytosine Biomarkers in Pancreatic Ductal Adenocarcinoma. Epigenomes 2019, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Ligation Sequencing DNA V14 (SQK-LSK114). Available online: https://nanoporetech.com/document/genomic-dna-by-ligation-sqk-lsk114 (accessed on 3 February 2025).
- Adaptive Sampling. Available online: https://nanoporetech.com/document/adaptive-sampling (accessed on 20 March 2025).
- de Bruin, D.D.S.H.; Haagmans, M.A.; van der Gaag, K.J.; Hoogenboom, J.; Weiler, N.E.C.; Tesi, N.; Salazar, A.; Zhang, Y.; Holstege, H.; Reinders, M.; et al. Exploring nanopore direct sequencing performance of forensic STRs, SNPs, InDels, and DNA methylation markers in a single assay. Forensic Sci. Int. Genet. 2025, 74, 103154. [Google Scholar] [CrossRef] [PubMed]
- Yuen, Z.W.S.; Shanmuganandam, S.; Stanley, M.; Jiang, S.; Hein, N.; Daniel, R.; McNevin, D.; Jack, C.; Eyras, E. Profiling age and body fluid DNA methylation markers using nanopore adaptive sampling. Forensic Sci. Int. Genet. 2024, 71, 103048. [Google Scholar] [CrossRef]
- Ciccarone, F.; Tagliatesta, S.; Caiafa, P.; Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms? Mech. Ageing Dev. 2018, 174, 3–17. [Google Scholar] [CrossRef]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; van de Voorde, W.; Decorte, R. Improved age determination of blood and teeth samples using a selected set of DNA methylation markers. Epigenetics 2015, 10, 922–930. [Google Scholar] [CrossRef]
- Freire-Aradas, A.; Phillips, C.; Mosquera-Miguel, A.; Girón-Santamaría, L.; Gómez-Tato, A.; Casares de Cal, M.; Álvarez-Dios, J.; Ansede-Bermejo, J.; Torres-Español, M.; Schneider, P.M.; et al. Development of a methylation marker set for forensic age estimation using analysis of public methylation data and the Agena Bioscience EpiTYPER system. Forensic Sci. Int. Genet. 2016, 24, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Lee, J.M.; Naue, J.; Fleckhaus, J.; Freire-Aradas, A.; Neubauer, J.; Pośpiech, E.; McCord, B.; Kalamara, V.; Gauthier, Q.; et al. A collaborative exercise on DNA methylation-based age prediction and body fluid typing. Forensic Sci. Int. Genet. 2022, 57, 102656. [Google Scholar] [CrossRef]
- Woźniak, A.; Heidegger, A.; Piniewska-Róg, D.; Pośpiech, E.; Xavier, C.; Pisarek, A.; Kartasińska, E.; Boroń, M.; Freire-Aradas, A.; Wojtas, M.; et al. Development of the VISAGE enhanced tool and statistical models for epigenetic age estimation in blood, buccal cells and bones. Aging 2021, 13, 6459–6484. [Google Scholar] [CrossRef]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Ż.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Naue, J.; Sänger, T.; Hoefsloot, H.C.J.; Lutz-Bonengel, S.; Kloosterman, A.D.; Verschure, P.J. Proof of concept study of age-dependent DNA methylation markers across different tissues by massive parallel sequencing. Forensic Sci. Int. Genet. 2018, 36, 152–159. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef]
- GenAge: Human Genes. Available online: https://genomics.senescence.info/genes/allgenes.php (accessed on 28 April 2025).
- Raj, K.; Horvath, S. Current perspectives on the cellular and molecular features of epigenetic ageing. Exp. Biol. Med. 2020, 245, 1532–1542. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Miyatake, S.; Lehtokari, V.L.; Todd, E.J.; Vornanen, P.; Yau, K.S.; Hayashi, Y.K.; Miyake, N.; Tsurusaki, Y.; Doi, H.; et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am. J. Hum. Genet. 2013, 93, 6–18. [Google Scholar] [CrossRef]
- Suguna, S.; Nandal, D.H.; Kamble, S.; Bharatha, A.; Kunkulol, R. Genomic DNA isolation from human whole blood samples by non enzymatic salting out method. Artic. Int. J. Pharm. Pharm. Sci. 2014, 6, 198–199. [Google Scholar]
- Nakamura, W.; Hirata, M.; Oda, S.; Chiba, K.; Okada, A.; Mateos, R.N.; Sugawa, M.; Iida, N.; Ushiama, M.; Tanabe, N.; et al. Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes. NPJ Genom. Med. 2024, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Adaptive Sampling Catalogue. Available online: https://community.nanoporetech.com/adaptive_sampling_catalogue (accessed on 3 February 2025).
- Fasta File for GRCh38. Available online: https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz (accessed on 3 February 2025).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ONT-CpG Site_Hg38 | IllmnID | Overlapped with | Slope | R2 | p-Value |
|---|---|---|---|---|---|
| chr1_1232655 | cg19945840 | Horvath353CpGs_hg18 | 0.0031 | 0.9303 | 0.0004 |
| chr10_35605501 | cg00168942 | Horvath353CpGs_hg18 | −0.0062 | 0.8008 | 0.0160 |
| chr10_48465490 | cg22796704 | Horvath391CpGs_hg19 | −0.0024 | 0.8664 | 0.0070 |
| Hannum71CpGs_hg18 | |||||
| chr15_31483691 | cg04875128 | Horvath391CpGs_hg19 | 0.0019 | 0.8017 | 0.0158 |
| Hannum71CpGs_hg18 | |||||
| chr15_51681722 | cg16717122 | Horvath391CpGs_hg19 | 0.0018 | 0.8589 | 0.0079 |
| chr16_66697409 | cg18693704 | Horvath391CpGs_hg19 | −0.0012 | 0.8488 | 0.0090 |
| chr20_46029585 | cg07547549 | Hannum71CpGs_hg18 | 0.0046 | 0.8376 | 0.0105 |
| Horvath391CpGs_hg19 | |||||
| chr6_30172367 | cg03771840 | Horvath391CpGs_hg19 | 0.0066 | 0.8947 | 0.0043 |
| chr9_34662284 | cg09722555 | Horvath353CpGs_hg18 | −0.0049 | 0.8165 | 0.0135 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Găitănaru, E.-C.; Popescu, R.G.; Stroe, A.-A.; Georgescu, S.E.; Marinescu, G.C. Exploring Epigenetic Ageing Using Direct Methylome Sequencing. Epigenomes 2025, 9, 25. https://doi.org/10.3390/epigenomes9030025
Găitănaru E-C, Popescu RG, Stroe A-A, Georgescu SE, Marinescu GC. Exploring Epigenetic Ageing Using Direct Methylome Sequencing. Epigenomes. 2025; 9(3):25. https://doi.org/10.3390/epigenomes9030025
Chicago/Turabian StyleGăitănaru, Elena-Cristina, Roua Gabriela Popescu, Andreea-Angelica Stroe, Sergiu Emil Georgescu, and George Cătălin Marinescu. 2025. "Exploring Epigenetic Ageing Using Direct Methylome Sequencing" Epigenomes 9, no. 3: 25. https://doi.org/10.3390/epigenomes9030025
APA StyleGăitănaru, E.-C., Popescu, R. G., Stroe, A.-A., Georgescu, S. E., & Marinescu, G. C. (2025). Exploring Epigenetic Ageing Using Direct Methylome Sequencing. Epigenomes, 9(3), 25. https://doi.org/10.3390/epigenomes9030025