
Association of Model-Predicted Epigenetic Age and Female Infertility

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
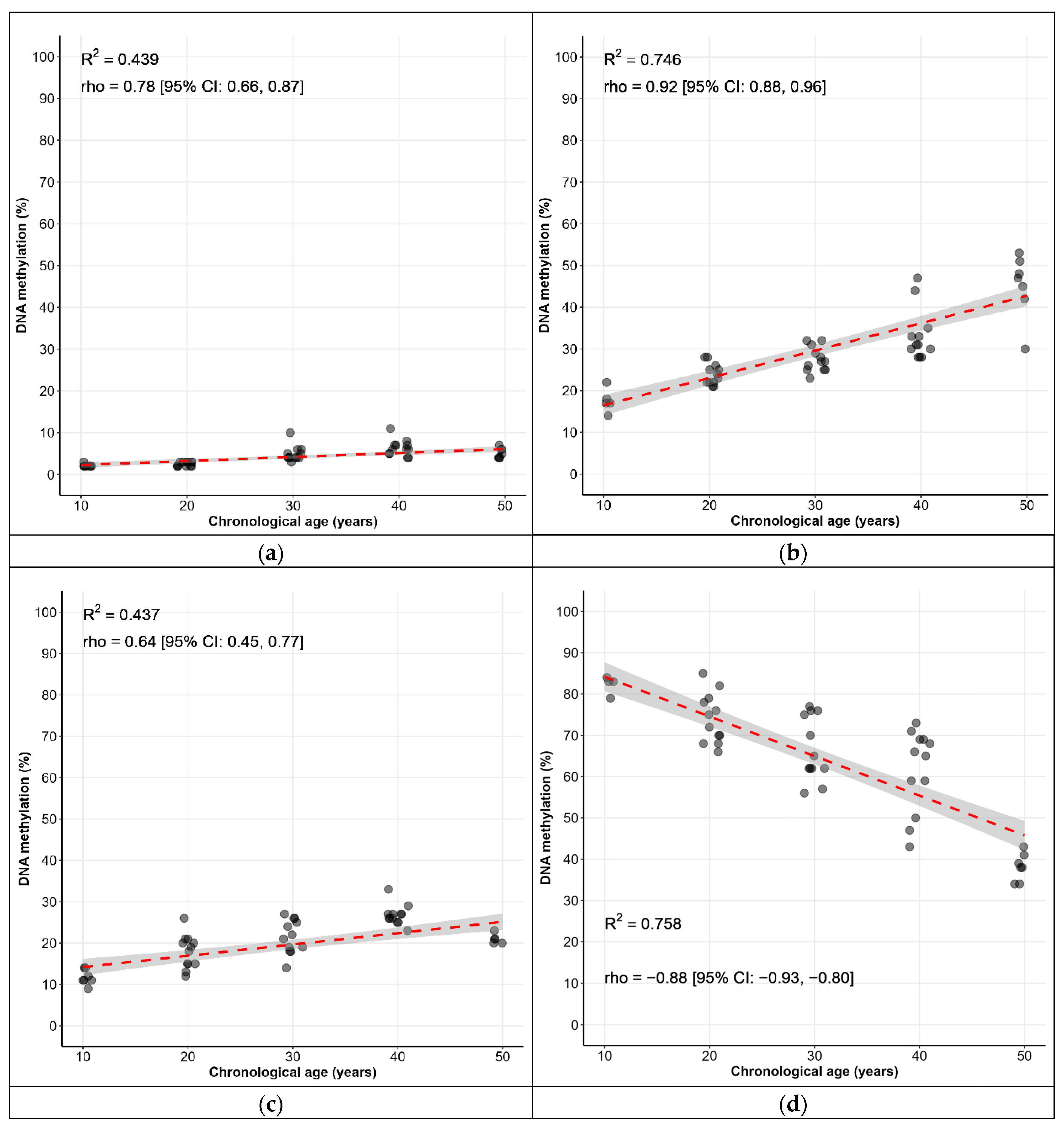
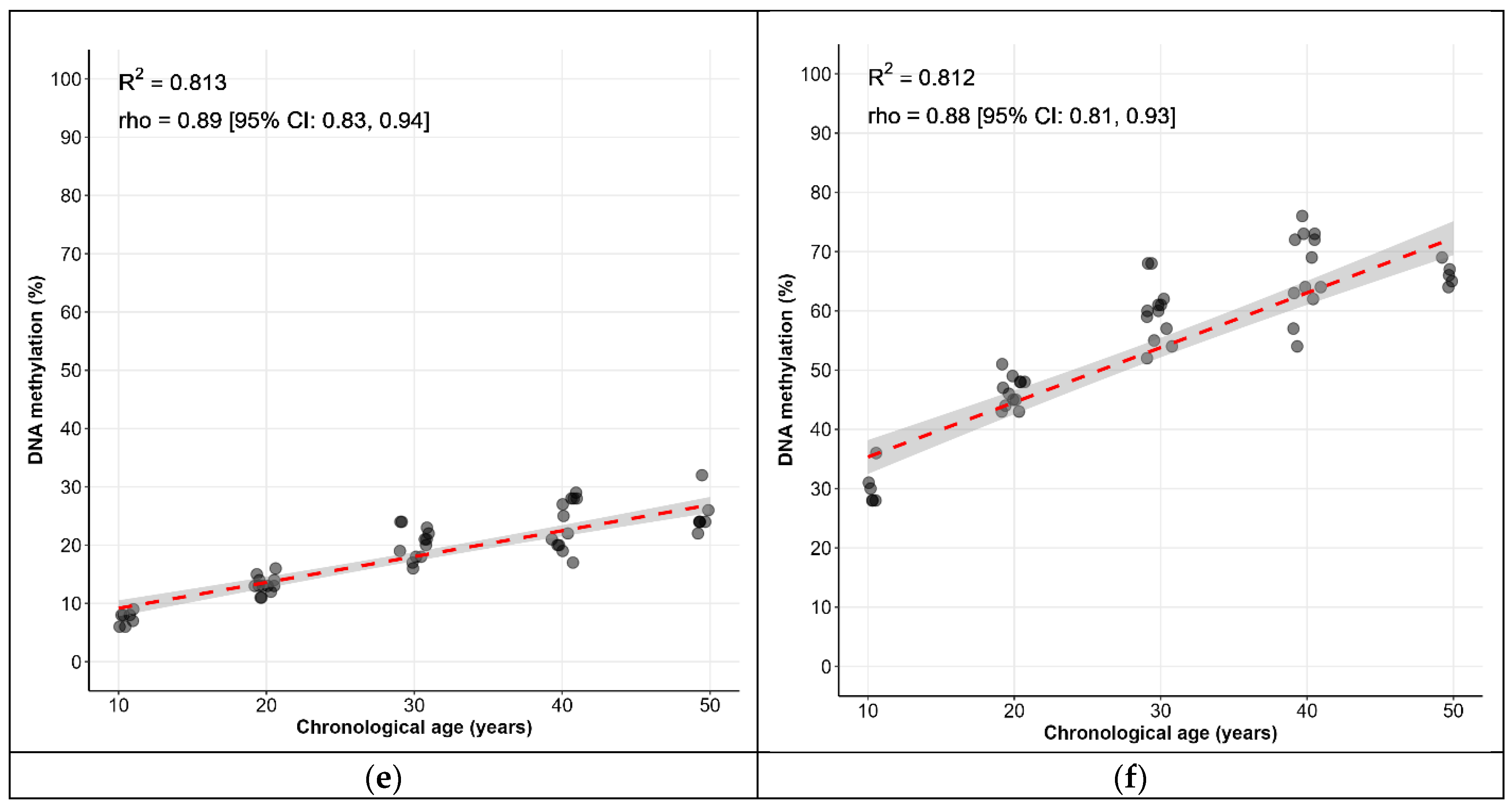
2.1. Selection of CpG Sites for Biological Age Prediction
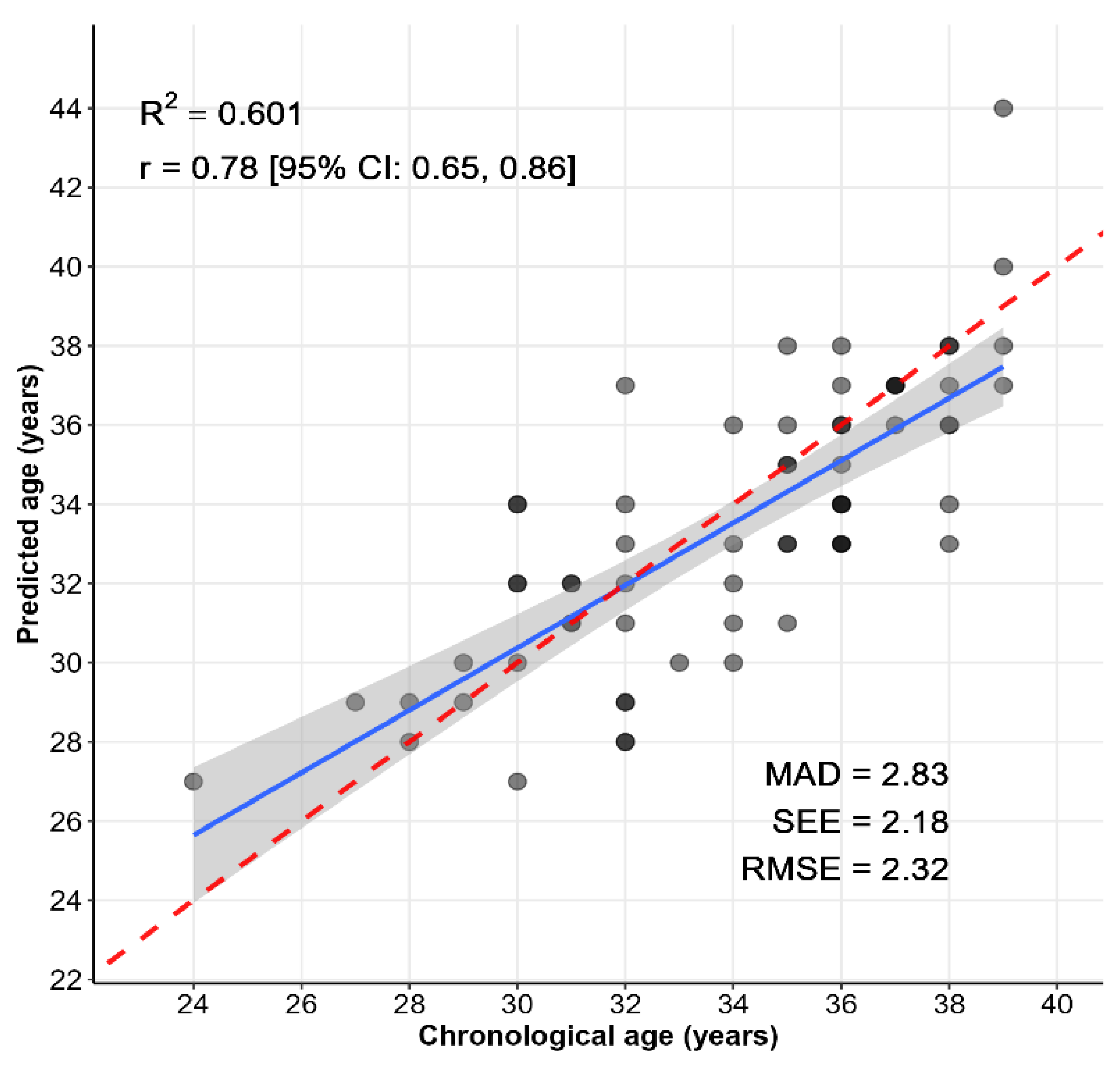
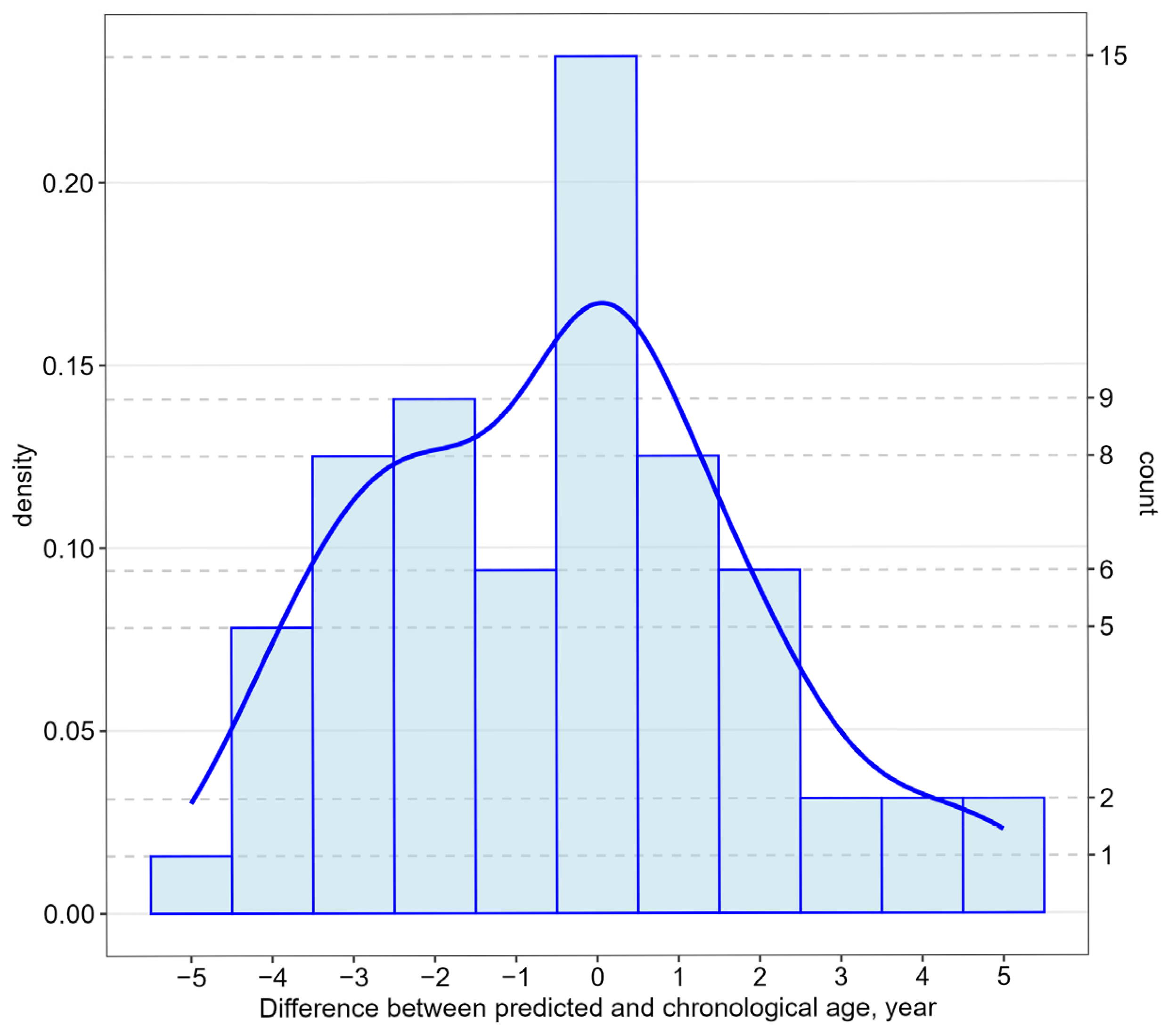
2.2. Biological Age Prediction Model
2.3. Testing of a Biological Age Calculation Model Using a Sample of Women with Perinatal Loss and Infertility
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. The Study Population and Blood Collection
4.3. DNA Extraction
4.4. Bisulfite Conversion
4.5. PCR and Pyrosequencing
4.6. Statistical Analysis
4.7. Development of the Age Prediction Model
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EAA | epigenetic age acceleration |
| EAD | epigenetic age deceleration |
| AMH | anti-Müllerian hormone |
| FSH | follicle-stimulating hormone |
| E2 | estradiol |
| AFC | antral follicle count |
| ART | assisted reproductive technology |
| BMI | body mass index |
| RIF | recurrent implantation failure |
| MAD | mean absolute deviation |
| MAE | mean absolute error |
| RMSE | root mean squared error |
| SEE | standard error of the estimate |
| SE | standard error |
| SD | standard deviation |
References
- Datta, J.; Palmer, M.J.; Tanton, C.; Gibson, L.J.; Jones, K.G.; Macdowall, W.; Glasier, A.; Sonnenberg, P.; Field, N.; Mercer, C.H.; et al. Prevalence of infertility and help seeking among 15 000 women and men. Hum. Reprod. 2016, 31, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Boedt, T.; Vanhove, A.C.; Vercoe, M.A.; Matthys, C.; Dancet, E.; Fong, S.L. Preconception lifestyle advice for people with infertility. Cochrane Database Syst. Rev. 2021, 4, CD008189. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, A.; Rogne, T.; Skara, K.H.; Haberg, S.E.; Page, C.M.; Fraser, A.; Burgess, S.; Lawlor, D.A.; Magnus, M.C. Body mass index and subfertility: Multivariable regression and Mendelian randomization analyses in the Norwegian Mother, Father and Child Cohort Study. Hum. Reprod. 2021, 36, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, A.; Wootton, R.E.; Page, C.M.; Skara, K.H.; Fraser, A.; Rogne, T.; Magnus, P.; Njolstad, P.R.; Andreassen, O.A.; Burgess, S.; et al. Smoking and infertility: Multivariable regression and Mendelian randomization analyses in the Norwegian Mother, Father and Child Cohort Study. Fertil. Steril. 2022, 118, 180–190. [Google Scholar] [CrossRef]
- ESHRE Capri Workshop Group. A prognosis-based approach to infertility: Understanding the role of time. Hum. Reprod. 2017, 32, 1556–1559. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef]
- Horvath SRaj, K. DNA methylation-based biomarkers and the epigenetic clock theory of aging. Nat. Rev.Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Zbiec-Piekarska, R.; Spolnicka, M.; Kupiec, T.; Makowska, Z.; Spas, A.; Parys-Proszek, A.; Kucharczyk, K.; Ploski, P.; Wojciech Branicki, W. Examination of DNA methylation status of the ELOVL2 marker may be useful for human age prediction in forensic science. Forensic Sci. Int. Genet. 2015, 14, 161–167. [Google Scholar] [CrossRef]
- Zbiec-Piekarska, R.; Spolnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Z.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Park, J.L.; Kim, J.H.; Seo, E.; Bae, D.H.; Kim, S.Y.; Lee, H.C.; Woo, K.M.; Kim, Y.S. Identification and evaluation of age-correlated DNA methylation markers for forensic use. Forensic Sci. Int. Genet. 2016, 23, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Jung, S.-E.; Hong, S.R.; Lee, E.H.; Lee, J.H.; Lee, S.D.; Lee, H.Y. Independent validation of DNA-based approaches for age prediction in blood. Forensic Sci. Int. Genet. 2017, 29, 250–256. [Google Scholar] [CrossRef]
- Spólnicka, M.; Pośpiech, E.; Pepłońska, B.; Zbieć-Piekarska, R.; Makowska, Ż.; Pięta, A.; Karłowska-Pik, J.; Ziemkiewicz, B.; Wężyk, M.; Gasperowicz, P.; et al. DNA methylation in ELOVL2 and C1orf132 correctly predicted chronological age of individuals from three disease groups. Int. J. Legal. Med. 2018, 132, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Spólnicka, M.; Pośpiech, E.; Adamczyk, J.G.; Freire-Aradas, A.; Pepłońska, B.; Zbieć-Piekarska, R.; Makowska, Ż.; Pięta, A.; Lareu, M.V.; Phillips, C.; et al. Modified aging of elite athletes revealed by analysis of epigenetic age markers. Aging 2018, 10, 241–252. [Google Scholar] [CrossRef]
- Marcante, B.; Marino, L.; Cattaneo, N.E.; Delicati, A.; Tozzo, P.; Caenazzo, L. Advancing forensic human chronological age estimation: Biochemical, genetic, and epigenetic approaches from the last 15 years: A systematic review. Int. J. Mol. Sci. 2025, 26, 3158. [Google Scholar] [CrossRef]
- Piani, L.L.; Vigano, P.; Somigliana, E. Epigenetic clocks and female fertility timeline: A new approach to one an old issue? Front. Cell Dev. Biol. 2023, 11, 1121231. [Google Scholar] [CrossRef]
- Shuster, L.T.; Gostout, B.S.; Grossardt, B.R.; Rocca, W.A. Prophylactic oophorectomy in premenopausal women and long-term health. Menopause Int. 2008, 14, 111–116. [Google Scholar] [CrossRef]
- Mason, J.B.; Cargill, S.L.; Anderson, G.B.; Carey, J.R. Transplantation of young ovaries to old mice increased life span in transplant recipients. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1207–1211. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Chen, B.H.; Hernandez, D.G.; Singleton, A.B.; Ferrucci, L.; Bandinelli, S.; Salfati, E.; Manson, J.E.; Quach, A.; et al. Menopause accelerates biological aging. Proc. Natl. Acad. Sci. USA 2016, 113, 9327–9332. [Google Scholar] [CrossRef]
- Xie, W.; Li, L.; Zheng, X.L.; Yin, W.D.; Tang, C.K. The role of Krüppel-like factor 14 in the pathogenesis of atherosclerosis. Atherosclerosis 2017, 263, 352–360. [Google Scholar] [CrossRef]
- Wu, S.; Wu, S.; Hsu, L.A.; Teng, M.S.; Chou, H.H.; Ko, Y.L. Differential genetic and epigenetic effects of the KLF14 gene on body shape indices and metabolic traits. Int. J. Mol. Sci. 2022, 23, 4165. [Google Scholar] [CrossRef]
- Koppes, E.; Shaffer, B.; Sadovsky, E.; Himes, K.; Barak, Y.; Sadovsky, Y.; Chaillet, J.R. Klf14 is an imprinted transcription factor that regulates placental growth. Placenta 2019, 88, 61–67. [Google Scholar] [CrossRef]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenet. Chromatin. 2018, 11, 25. [Google Scholar] [CrossRef]
- Zhu, T.; Zheng, S.C.; Paul, D.S.; Horvath, S.; Teschendorff, A.E. Cell and tissue type independent age-associated DNA methylation changes are not rare but common. Aging 2018, 10, 3541–3557. [Google Scholar] [CrossRef]
- Ibáñez-Cabellos, J.S.; Sandoval, J.; Pallardó, F.V.; García-Giménez, J.L.; Mena-Molla, S. A sex-specific minimal CpG-based model for biological aging using ELOVL2 methylation analysis. Int. J. Mol. Sci. 2025, 26, 392. [Google Scholar] [CrossRef]
- Chen, D.; Chao, D.L.; Rocha, L.; Kolar, M.; Nguyen Huu, V.A.; Krawczyk, M.; Dasyani, M.; Wang, T.; Jafari, M.; Jabari, M.; et al. The lipid elongation enzyme ELOVL2 is a molecular regulator of aging in the retina. Aging Cell 2020, 19, e13100. [Google Scholar] [CrossRef]
- Chao, D.L.; Skowronska-Krawczyk, D. ELOVL2: Not just a biomarker of aging. Transl. Med. Aging 2020, 4, 78–80. [Google Scholar] [CrossRef]
- Durso, D.F.; Bacalini, M.G.; Sala, C.; Pirazzini, C.; Marasco, E.; Bonafé, M.; do Valle, Í.F.; Gentilini, D.; Castellani, G.; Faria, A.M.C.; et al. Acceleration of leukocytes’ epigenetic age as an early tumor and sex-specific marker of breast and colorectal cancer. Oncotarget 2017, 8, 23237–23245. [Google Scholar] [CrossRef]
- González, R.S.; Rodriguez-Cruz, M.; Maldonado, J.; Saavedra, F.J. Role of maternal tissue in the synthesis of polyunsaturated fatty acids in response to a lipid-deficient diet during pregnancy and lactation in rats. Gene 2014, 549, 7–23. [Google Scholar] [CrossRef]
- Jung, S.E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef]
- Lee, H.Y.; Hong, S.R.; Lee, J.E.; Hwang, I.K.; Kim, N.Y.; Lee, J.M.; Fleckhaus, J.; Jung, S.E.; Lee, Y.H. Epigenetic age signatures in bones. Forensic Sci. Int. Genet. 2020, 46, 102261. [Google Scholar] [CrossRef]
- Habibe, J.J.; Clemente-Olivo, M.P.; Vries, C.J. How (Epi)genetic regulation of the LIM-domain protein FHL2 impacts multifactorial disease. Cells 2021, 10, 2611. [Google Scholar] [CrossRef]
- Bae, H.; Lunetta, K.L.; Murabito, J.M.; Andersen, S.L.; Schupf, N.; Perls, T.; Sebastiani, P. Genetic associations with age of menopause in familial longevity. Menopause 2019, 26, 1204–1212. [Google Scholar] [CrossRef]
- Tachmazidou, I.; Süveges, D.; Min, J.L.; Ritchie, G.R.S.; Steinberg, J.; Walter, K.; Iotchkova, V.; Schwartzentruber, J.; Huang, J.; Memari, Y.; et al. Whole-genome sequencing coupled to imputation discovers genetic signals for anthropometric traits. Am. J. Hum. Genet. 2017, 100, 865–884. [Google Scholar] [CrossRef]
- Zhu, Z.; Guo, Y.; Shi, H.; Liu, C.L.; Panganiban, R.A.; Chung, W.; O’Connor, L.J.; Himes, B.E.; Gazal, S.; Hasegawa, K.; et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. J. Allergy Clin. Immunol. 2020, 145, 537–549. [Google Scholar] [CrossRef]
- Shafaroudi, A.M.; Sharifi-Zarchi, A.; Saeid Rahmani, S.; Nafissi, N.; Mowla, S.J.; Lauria, A.; Oliviero, S.; Matin, M.M. Expression and function of C1orf132 long-noncoding RNA in breast cancer cell lines and tissues. Int. J. Mol. Sci. 2021, 22, 6768. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, L.; Yu, Y.; Li, D.; Zhu, X.; Yan, D.; Zhu, Z. TRIM59: A potential diagnostic and prognostic biomarker in human tumors. PLoS ONE 2021, 16, e0257445. [Google Scholar] [CrossRef]
- Wang, F.; Wang, H.; Sun, L.; Niu, C.; Xu, J. TRIM59 inhibits PPM1A through ubiquitination and activates TGF-β/Smad signaling to promote the invasion of ectopic endometrial stromal cells in endometriosis. Am. J. Physiol. Cell Physiol. 2020, 319, C392–C401. [Google Scholar] [CrossRef]
- Wu, T.; Zhou, H.; Wang, L.; Tan, J.; Gao, W.; Wu, Y.; Zhao, D.; Shen, C.; Zheng, B.; Huang, X.; et al. TRIM59 is required for mouse GC-1 cell maintenance through modulating the ubiquitination of AXIN1. Heliyon 2024, 10, e36744. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench. Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques”, 4th ed.; Morgan Kaufmann: Burlington, NJ, USA, 2016. [Google Scholar]
- Fritz, R.; Jindal, S. Reproductive aging and elective fertility preservation. J. Ovarian Res. 2018, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Cil, A.P.; Turkgeldi, L.; Seli, E. Oocyte cryopreservation as a preventive measure for age-related fertility loss. Semin. Reprod. Med. 2015, 33, 429–435. [Google Scholar] [CrossRef]
- Daunay, A.; Baudrin, L.G.; Deleuze, J.F.; How-Kit, A. Evaluation of six blood-based age prediction models using DNA methylation analysis by pyrosequencing. Sci. Rep. 2019, 9, 8862. [Google Scholar] [CrossRef] [PubMed]
- Thong, Z.; Liang Shun Chan, X.; Ying Ying Tan, J.; Shuzhen Loo, E.; Kiu Choong Syn, C. Evaluation of DNA methylation-based age prediction on blood. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e249–e251. [Google Scholar] [CrossRef]
- Hanson, B.M.; Tao, X.; Zhan, Y.; Jenkins, T.G.; Morin, S.J.; Scott, R.T.; Seli, E.U. Young women with poor ovarian response exhibit epigenetic age acceleration based on evaluation of white blood cells using a DNA methylation-derived age prediction model. Hum. Reprod. 2020, 35, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Monseur, B.; Murugappan, G.; Bentley, J.; Teng, N.; Westphal, L. Epigenetic clock measuring age acceleration via DNA methylation levels in blood is associated with decreased oocyte yield. J. Assist. Reprod. Genet. 2020, 37, 1097–1103. [Google Scholar] [CrossRef]
- Tal, R.; Seifer, D.B. Ovarian reserve testing: A user’s guide. Am. J. Obstet. Gynecol. 2017, 217, 129–140. [Google Scholar] [CrossRef]
- Dogan, M.V.; Xiang, J.; Beach, S.R.H.; Cutrona, C.; Gibbons, F.X.; Simons, R.L.; Brody, G.H.; Stapleton, J.T.; Philibert, R.A. Ethnicity and smoking-associated DNA methilation changes at HIV co-receptor GPR15. Front. Pshychiatry 2015, 6, 132. [Google Scholar] [CrossRef]
- Lei, M.K.; Gibbons, F.X.; Simons, R.L.; Philibert, R.A.; Beach, S.R.H. The Effect of Tobacco Smoking Differs across Indices of DNA Methylation-Based Aging in an African American Sample: DNA Methylation-Based Indices of Smoking Capture These Effects. Genes 2020, 11, 311. [Google Scholar] [CrossRef]
- Philibert, R.; Beach, S.R.H.; Lei, M.K.; Gibbons, F.X.; Gerrard, M.; Simons, R.L.; Dogan, M.V. Array-Based Epigenetic Aging Indices May Be Racially Biased. Genes 2020, 11, 685. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.; Bandinelli, S.; Hou, L.; Baccarelli, A.; Stewart, J.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed]
- Oblak, L.; van der Zaag, J.; Higgins-Chen, A.T.; Levine, M.E.; Boks, M.P. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Res. Rev. 2021, 69, 101348. [Google Scholar] [CrossRef]
- Rodés, B.; Cadiñanos, J.; Esteban-Cantos, A.; Rodríguez-Centeno, J.; Ramón Arribas, J. Ageing with HIV: Challenges and biomarkers. eBioMedicine 2022, 77, 103896. [Google Scholar] [CrossRef]
- Ramaker, M.E.; Corcoran, D.L.; Apsley, A.T.; Kobor, M.S.; Kraus, V.B.; Kraus, W.E.; Lin, D.T.S.; Orenduff, M.C.; Pieper, C.F.; Waziry, R.; et al. Epigenome-wide Association Study Analysis of Calorie Restriction in Humans, CALERIETM Trial Analysis. J. Gerontol. A. Biol. Sci. Med. Sci. 2022, 77, 2395–2401. [Google Scholar] [CrossRef]
- Cao, X.; Li, W.; Wang, T.; Ran, D.; Davalos, V.; Planas-Serra, L.; Pujol, A.; Esteller, M.; Wang, X.; Yu, H. Accelerated biological aging in COVID-19 patients. Nat. Commun. 2022, 13, 2135. [Google Scholar] [CrossRef] [PubMed]
- Moqri, M.; Herzog, C.; Poganik, J.R.; Ying, K.; Justice, J.N.; Belsky, D.W.; Higgins-Chen, A.T.; Chen, B.H.; Cohen, A.A.; Fuellen, G.; et al. Validation of biomarkers of aging. Nat. Med. 2024, 30, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimA.e strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Krolevets, M.; Cate, V.T.; Prochaska, J.H.; Schulz, A.; Rapp, S.; Tenzer, S.; Andrade-Navarro, M.A.; Horvath, S.; Niehrs, C.; Wild, P.S. DNA methylation and cardiovascular disease in humans: A systematic review and database of known CpG methylation sites. Clin. Epigenetics 2023, 15, 56. [Google Scholar] [CrossRef]
- Li, A.; Koch, Z.; Ideker, T. Epigenetic aging: Biological age prediction and informing amechanistic theory of aging. J. Intern. Med. 2022, 292, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.r-project.org/ (accessed on 27 October 2024).
- Wickham, H. ggplot2. Elegant Graphics for Data Analysis, 2nd ed.; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Patil, I. Visualizations with statistical details: The ‘ggstatsplot’ approach. J. Open Source Softw. 2021, 6, 3167. [Google Scholar] [CrossRef]
- Lüdecke, D.; Ben-Shachar, M.S.; Patil, I.; Waggoner, P.; Makowski, D. Performance: An R Package for Assessment, Comparison and Testing of Statistical Models. J. Open Source Softw. 2021, 6, 3139. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | CpG Sites | Spearman’s ρ [95% CI] * |
|---|---|---|
| KLF14 | C1 | 0.77 [0.67, 0.84] |
| FHL2 | C2 | 0.85 [0.79, 0.90] |
| TRIM59 | C7 | 0.60 [0.45, 0.72] |
| C1orf132 | C1 | −0.92 [−0.95, −0.88] |
| ELOVL2 | C5 | 0.94 [0.91, 0.96] |
| C7 | 0.88 [0.82, 0.92] |
| Age Group, Year (n) | Methylation %, Mean (SD) * | |||||
|---|---|---|---|---|---|---|
| KLF14 | FHL2 | TRIM59 | C1orf132 | ELOVL2 C5 | ELOVL2 C7 | |
| 10 (12) | 2.17 (0.39) | 17.7 (2.46) | 11.4 (1.62) | 82.2 (2.52) | 8.08 (1.51) | 30.1 (2.43) |
| 20 (12) | 2.50 (0.52) ↑ | 23.9 (2.50) ↑ | 17.9 (4.03) ↑ | 74.1 (6.05) ↓ | 13.2 (1.47) ↑ | 46.4 (2.50) ↑ |
| 30 (12) | 4.92 (1.83) ↑ | 27.5 (2.97) ↑ | 21.6 (4.08) ↑ | 66.7 (7.74) ↓ | 20.2 (2.70) ↑ | 59.8 (4.94) ↑ |
| 40 (12) | 6.33 (1.92) ↑ | 33.2 (6.19) ↑ | 26.8 (2.45) ↑ | 61.6 (10.0) ↓ | 23.7 (4.27) ↑ | 66.6 (6.96) ↑ |
| 50 (12) | 5.00 (1.04) ↓ | 45.8 (8.53) ↑ | 20.7 (2.77) ↓ | 40.4 (6.08) ↓ | 25.0 (2.63) ↑ | 66.2 (1.99) ↓ |
| 60 (12) | 6.50 (1.24) ↑ | 37.8 (4.45) ↓ | 23.5 (3.29) ↑ | 38.1 (6.04) ↓ | 30.7 (3.23) ↑ | 72.8 (3.16) ↑ |
| 70 (12) | 7.58 (2.43) ↑ | 45.8 (7.19) ↑ | 26.1 (10.4) ↑ | 28.7 (10.1) ↓ | 37.6 (8.37) ↑ | 75.6 (3.55) ↑ |
| 80 (10) | 7.70 (2.21) ↑ | 46.6 (5.10) ↑ | 30.1 (6.06) ↑ | 29.2 (7.15) ↑ | 37.0 (5.25) ↓ | 72.1 (2.81) ↓ |
| Name | Model Equation | SE | Statistical Summary |
|---|---|---|---|
| Age_predict1 | Age = 24.4506 + 0.1492 × FHL2 + 0.4488 × KLF14 + 0.0114 × TRIM59 + −0.3771 × C1orf132 + 0.3328 × ELOVL2 C5 + 0.3259 × ELOVL2 C7 | 5.727 0.0833 0.3094 0.1432 0.0514 0.2235 0.1148 | R2: 0.954 R2Adjusted: 0.9488 Correlation coefficient: 0.9674 MAE: 2.6158 RMSE: 3.5943 Relative absolute error: 21.42% Root relative squared error: 25.17% AIC: 319 |
| Age_predict2 | Age = 24.3118 + 0.1495 × FHL2 + 0.4492 × KLF14 + −0.3755 × C1orf132 + 0.3335 × ELOVL2 C5 + 0.3303 × ELOVL2 C7 | 5.4046 0.0825 0.3065 0.047 0.2212 0.0996 | R2: 0.954 R2Adjusted: 0.94975 Correlation coefficient: 0.9675 MAE: 2.6039 RMSE: 3.5866 Relative absolute error: 21.33% Root relative squared error: 25.12% AIC: 317 |
| Group | n | Age, y (SD) | BMI, kg/m2 (SD) | AMH, ng/mL (SD) |
|---|---|---|---|---|
| Group I. Control group: healthy individuals who have had at least one live birth and have no history of perinatal losses or infertility diagnoses. | 7 | 33.3 (3.5) | 21 (3.7) | 5.2 (3.2) |
| Group II. Women with a history of infertility diagnosis or perinatal losses; ART was not applied. | 16 | 33.4 (3.4) | 21 (1.5) | 3.6 (4.1) |
| Group III. Women experiencing infertility or perinatal losses and have undergone ART and achieved successful pregnancies, resulting in delivery. | 29 | 34.1 (3.6) | 22 (3.3) | 2.1 (1.5) |
| Group IV. Women experiencing infertility, perinatal losses, and assisted reproductive technology (ART), without pregnancy occurrence. | 12 | 34.6 (3.1) | 22 (4.2) | 3.4 (1.4) |
| n (%) | ||||
| Age: | ||||
| 24–29 | 6 (9%) | |||
| 30–34 | 25 (39%) | |||
| 35–39 | 33 (52%) | |||
| BMI abnormal (>18.5 or <25) | 16 (25%) | |||
| AMH < 1.2 ng/mL | 17 (27%) | |||
| Group | n | Chronological Age, y | Predicted Age, y | Statistical Significance of Differences Between Chronological and Predicted Age p | EAA/EAD (Differences Between Actual and Predicted Ages) | |||
|---|---|---|---|---|---|---|---|---|
| Median | p * | Median | p | Median | p | |||
| Group I Group II Group III Group IV | 7 16 29 12 | 33.00 33.00 35.00 35.50 | 0.73 | 32.80 33.35 33.30 33.50 | 0.76 | 0.67 0.53 0.30 0.56 | 0.00 −0.15 0.60 0.10 | 0.99 |
| ART: yes no | 41 23 | 35.00 33.00 | 0.28 | 33.30 33.20 | 0.33 | 0.19 0.37 | 0.50 −0.10 | 0.86 |
| BMI, kg/m2: normal deviation | 48 16 | 35.00 33.50 | 0.31 | 33.40 32.50 | 0.19 | 0.31 0.19 | 0.40 0.20 | 0.54 |
| AMH, ng/mL: >1.2 <1.2 | 47 17 | 33.00 35.00 | 0.19 | 33.20 33.30 | 0.26 | 0.47 0.08 | −0.10 1.70 | 0.35 |
| Locus | Gene Name | Gene Function | CpG Sites | Chromosome Location (GRCh38) |
|---|---|---|---|---|
| KLF14 | Kruppel-like factor 14 | Transcription factor | C1 | Chr7: 130734355 |
| FHL2 | Four and a half LIM domains protein 2 | Transcription factor | C2 | Chr2: 105399288 |
| TRIM59 | Tripartite motif containing 59 | Regulator of immune signaling pathways | C7 | Chr3: 160450199 |
| C1orf132 | - | - | C1 | Chr1: 207823681 |
| ELOVL2 | ELOVL fatty acid elongase 2 | Synthesis of very long chain polyunsaturated fatty acids | C5 | Chr6: 11044875 |
| C7 | Chr6: 11044867 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozdysheva, E.; Korchagin, V.; Rumyantseva, T.; Ogneva, D.; Zhivotova, V.; Gaponova, I.; Mironov, K.; Akimkin, V. Association of Model-Predicted Epigenetic Age and Female Infertility. Epigenomes 2025, 9, 19. https://doi.org/10.3390/epigenomes9020019
Pozdysheva E, Korchagin V, Rumyantseva T, Ogneva D, Zhivotova V, Gaponova I, Mironov K, Akimkin V. Association of Model-Predicted Epigenetic Age and Female Infertility. Epigenomes. 2025; 9(2):19. https://doi.org/10.3390/epigenomes9020019
Chicago/Turabian StylePozdysheva, Elena, Vitaly Korchagin, Tatiana Rumyantseva, Daria Ogneva, Vera Zhivotova, Irina Gaponova, Konstantin Mironov, and Vasily Akimkin. 2025. "Association of Model-Predicted Epigenetic Age and Female Infertility" Epigenomes 9, no. 2: 19. https://doi.org/10.3390/epigenomes9020019
APA StylePozdysheva, E., Korchagin, V., Rumyantseva, T., Ogneva, D., Zhivotova, V., Gaponova, I., Mironov, K., & Akimkin, V. (2025). Association of Model-Predicted Epigenetic Age and Female Infertility. Epigenomes, 9(2), 19. https://doi.org/10.3390/epigenomes9020019