

Novel Epigenetics Control (EpC) Nanocarrier for Cancer Therapy Through Dual-Targeting Approach to DNA Methyltransferase and Ten-Eleven Translocation Enzymes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
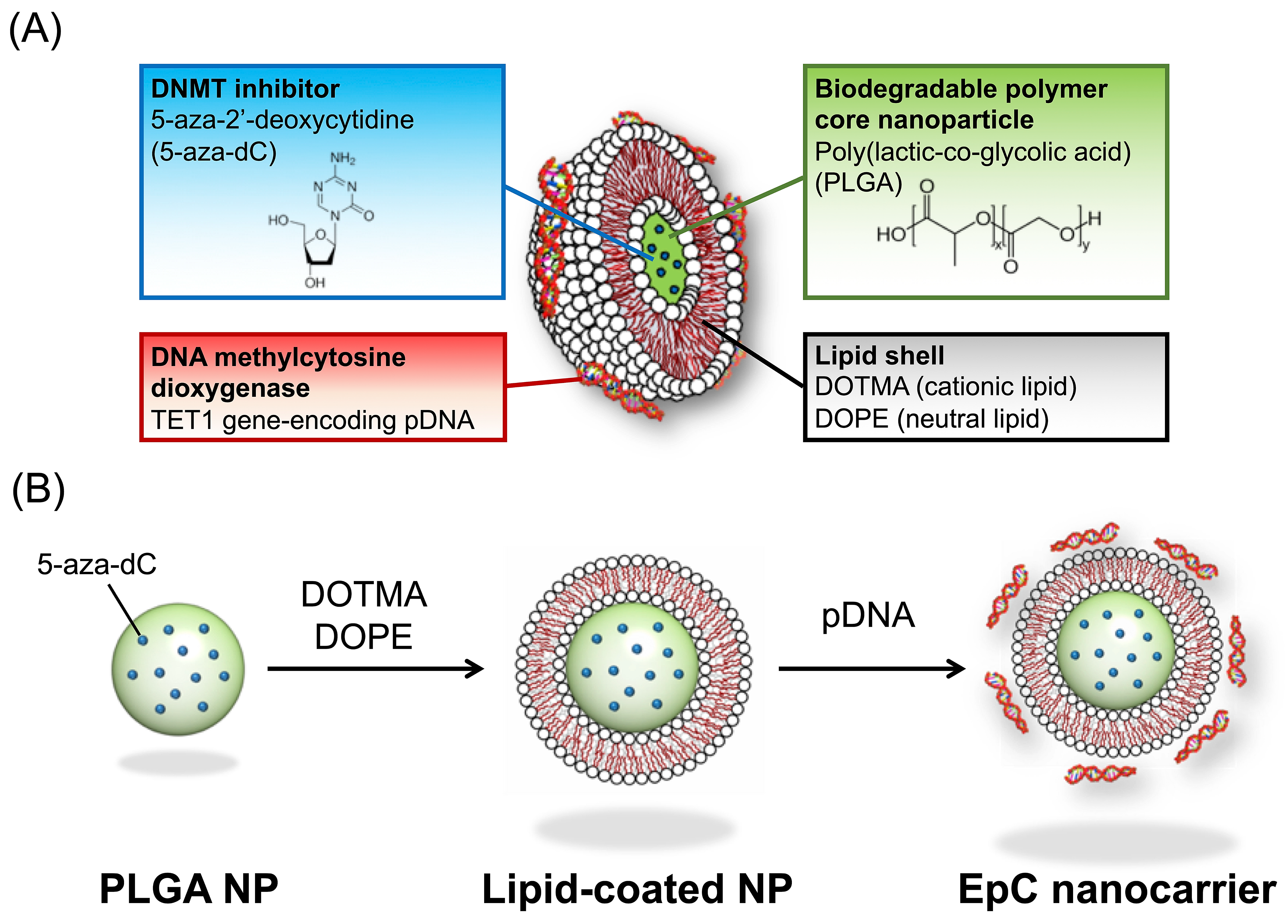
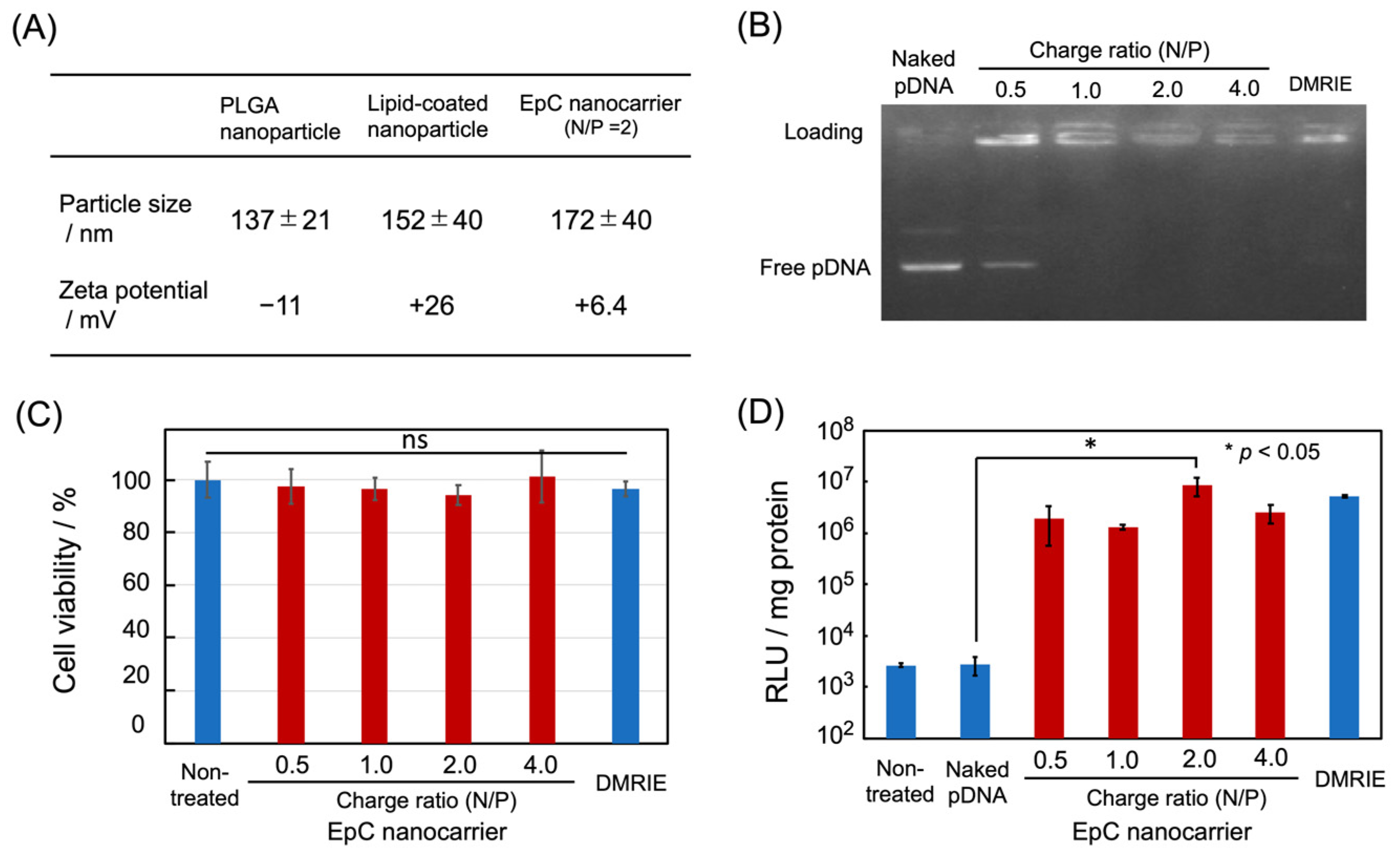
2.1. Preparation of EpC Nanocarrier
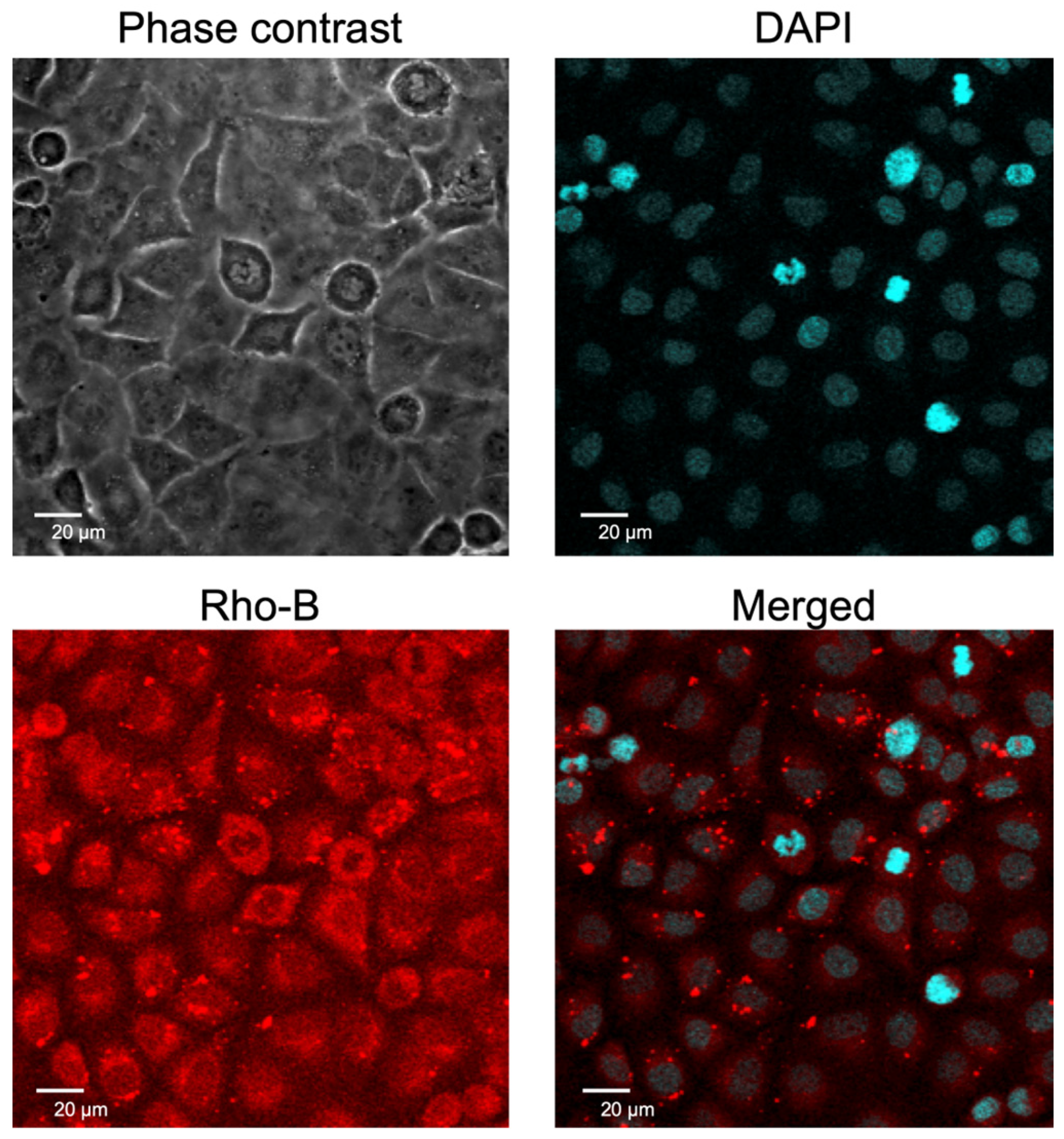
2.2. Cellular Uptake of EpC Nanocarrier
2.3. Upregulation of p53 Protein in HCT116 Cells
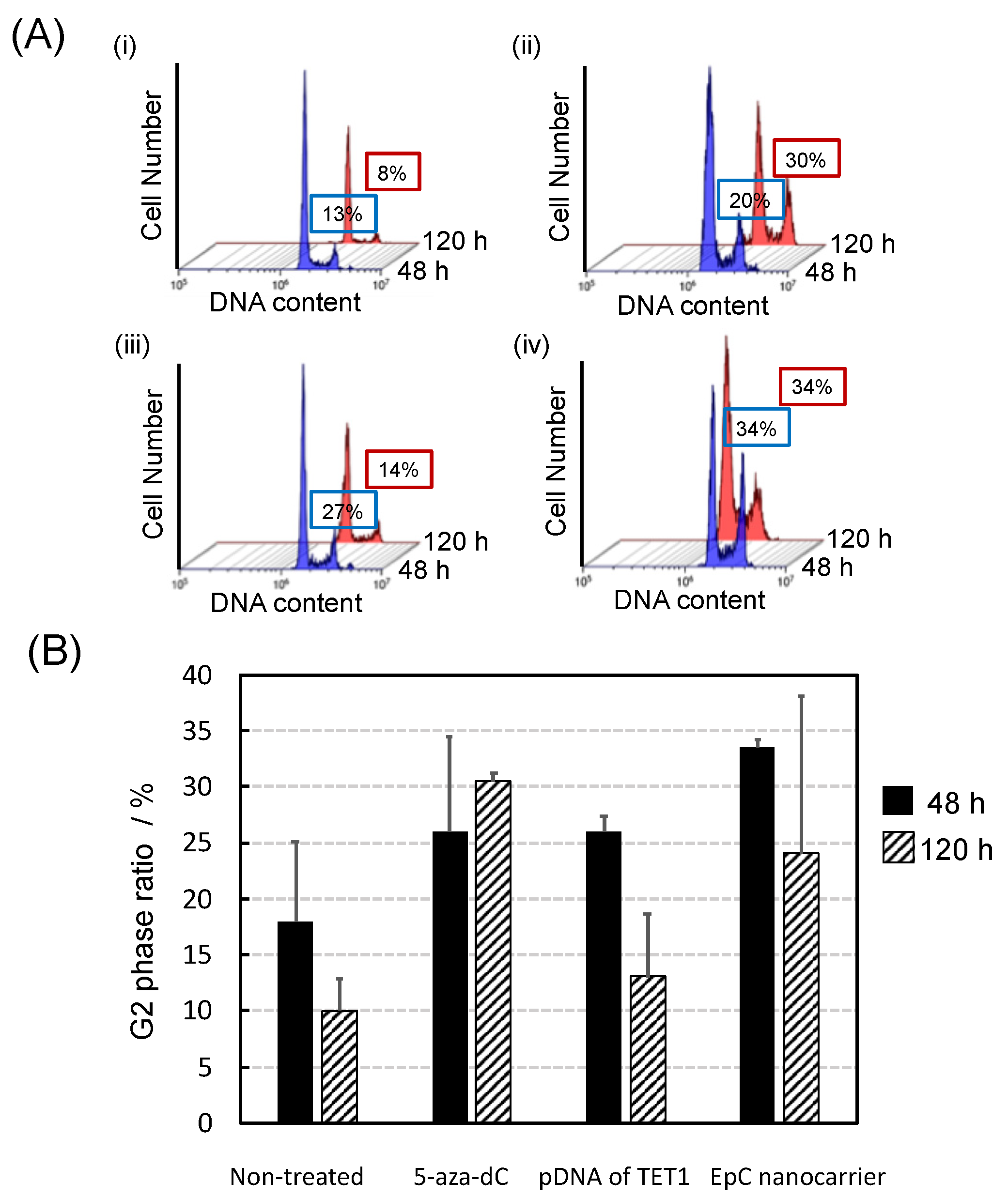
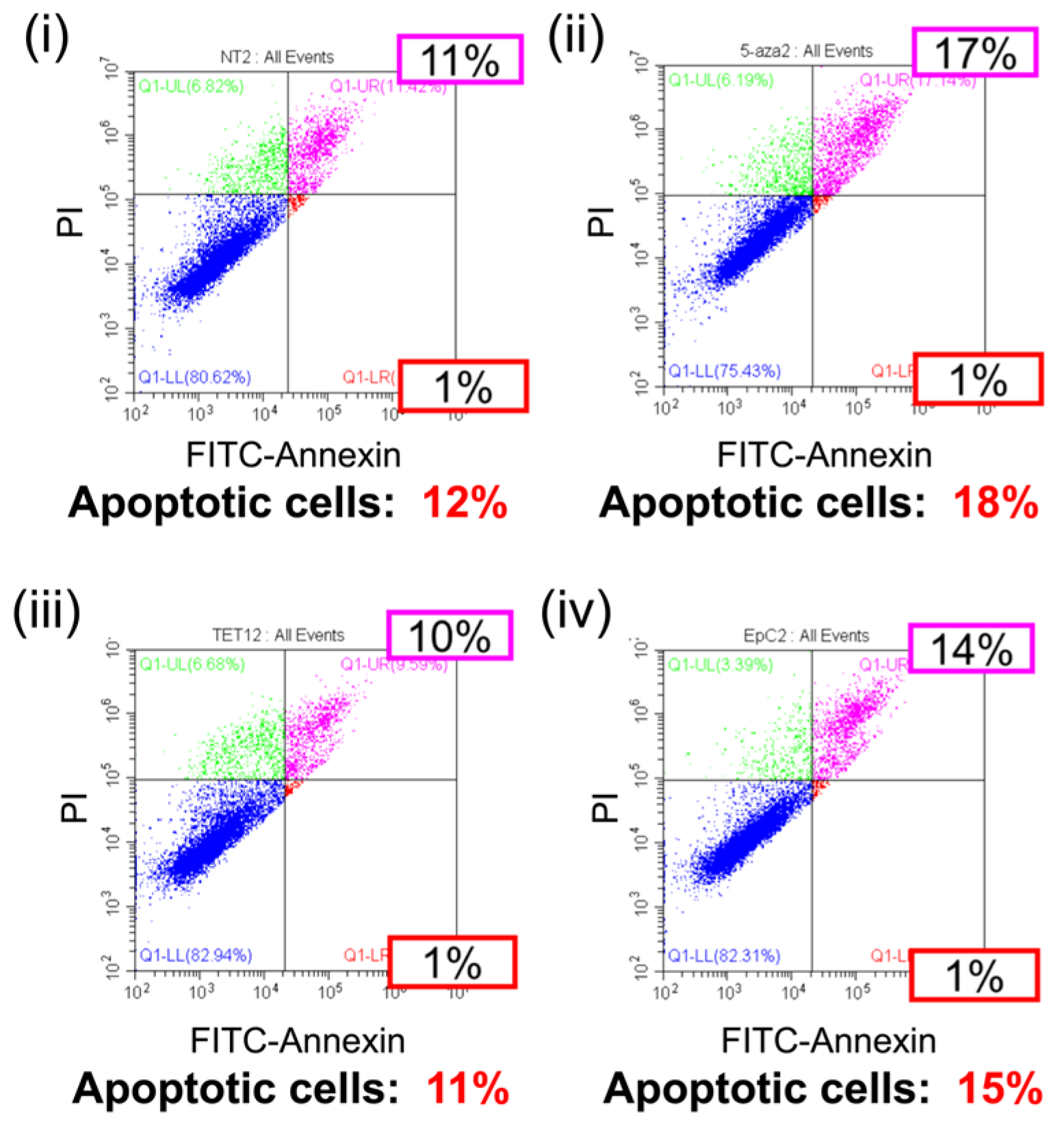
2.4. Anti-Tumor Effects of EpC Nanocarrier
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Preparation of EpC Nanocarrier
4.3. Agarose Gel Electrophoresis
4.4. Luciferase Assay
4.5. Alamar Blue Assay
4.6. Intracellular Localization
4.7. Western Blotting
4.8. Cell Cycle Analysis
4.9. Apoptosis Assay
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, X.; Zhao, H.; Wang, R.; Chen, Y.; Ouyang, X.; Li, W.; Sun, Y.; Peng, A. Cancer epigenetics: From laboratory studies and clinical trials to precision medicine. Cell Death Discov. 2024, 10, 28. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef]
- Kang, J.H.; Kim, S.J.; Noh, D.Y.; Park, I.A.; Choe, K.J.; Yoo, O.J.; Kang, H.S. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: Correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab. Investig. 2001, 81, 573–579. [Google Scholar] [CrossRef]
- Petrovich, M.; Veprintsev, D.B. Effects of CpG methylation on recognition of DNA by the tumour suppressor p53. J. Mol. Biol. 2009, 386, 72–80. [Google Scholar] [CrossRef]
- Kagan, A.B.; Garrison, D.A.; Anders, N.M.; Webster, J.A.; Baker, S.D.; Yegnasubramanian, S.; Rudek, M.A. DNA methyltransferase inhibitor exposure-response: Challenges and opportunities. Clin. Transl. Sci. 2023, 16, 1309–1322. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, G.; Li, Y.; Lei, D.; Xiang, J.; Ouyang, L.; Wang, Y.; Yang, J. Recent progress in DNA methyltransferase inhibitors as anticancer agents. Front. Pharmacol. 2022, 13, 1072651. [Google Scholar] [CrossRef]
- Shi, M.Q.; Xu, Y.; Fu, X.; Pan, D.S.; Lu, X.P.; Xiao, Y.; Jiang, Y.Z. Advances in targeting histone deacetylase for treatment of solid tumors. J. Hematol. Oncol. 2024, 17, 37. [Google Scholar] [CrossRef]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Tao, L.; Zhou, Y.; Luo, Y.; Qiu, J.; Xiao, Y.; Zou, J.; Zhang, Y.; Liu, X.; Yang, X.; Gou, K.; et al. Epigenetic regulation in cancer therapy: From mechanisms to clinical advances. MedComm-Oncology 2024, 3, e59. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Wang, C.; Wang, X. TET (Ten-eleven translocation) family proteins: Structure, biological functions and applications. Signal Transduct. Target. Ther. 2023, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Panjarian, S.; Issa, J.J. The Roles of DNA Demethylases in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 628. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, K.; Arima, K.; Asaba, Y.; Kubota, R.; Asayama, S.; Sato, K.; Kawakami, H. The potential of lipid-polymer nanoparticles as epigenetic and ROS control approaches for COPD. Free Radic. Res. 2020, 54, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Arima, K.; Kubota, R.; Sato, K.; Kawakami, H. Preparation of Mitochondria- and Epigenetics-Targeting Nanoparticles for Suppression of Cancer Metastasis. Part. Part. Syst. Charact. 2021, 38, 2100003. [Google Scholar] [CrossRef]
- Essa, D.; Kondiah, P.P.D.; Choonara, Y.E.; Pillay, V. The Design of Poly(lactide-co-glycolide) Nanocarriers for Medical Applications. Front. Bioeng. Biotechnol. 2020, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yan, Y.; Yu, W.; Zhang, H. Role of ten-eleven translocation proteins and 5-hydroxymethylcytosine in hepatocellular carcinoma. Cell Prolif. 2019, 52, e12626. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Liu, S.; Breslin, S.J.P.; Zhang, J. Mechanisms that regulate the activities of TET proteins. Cell. Mol. Life Sci. 2022, 79, 363. [Google Scholar] [CrossRef]
- Sajadian, S.O.; Tripura, C.; Samani, F.S.; Ruoss, M.; Dooley, S.; Baharvand, H.; Nussler, A.K. Vitamin C enhances epigenetic modifications induced by 5-azacytidine and cell cycle arrest in the hepatocellular carcinoma cell lines HLE and Huh7. Clin. Epigenetics 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jung, I.; Lee, C.H.; An, J.; Ko, M. Development of Novel Epigenetic Anti-Cancer Therapy Targeting TET Proteins. Int. J. Mol. Sci. 2023, 24, 16375. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Mao, S.Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef] [PubMed]
- Dickson, K.M.; Gustafson, C.B.; Young, J.I.; Züchner, S.; Wang, G. Ascorbate-induced generation of 5-hydroxymethylcytosine is unaffected by varying levels of iron and 2-oxoglutarate. Biochem. Biophys. Res. Commun. 2013, 439, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Palii, S.S.; Van Emburgh, B.O.; Sankpal, U.T.; Brown, K.D.; Robertson, K.D. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 2008, 28, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, J.; Sidransky, D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 2010, 29, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Tomkova, M.; Schuster-Böckler, B. DNA Modifications: Naturally More Error Prone? Trends Genet. 2018, 34, 627–638. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Mao, H.; Du, Z.; Chan, W.Y.; Murray, P.; Luo, B.; Chan, A.T.; Mok, T.S.; Chan, F.K.; et al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Sci. Rep. 2016, 6, 26591. [Google Scholar] [CrossRef]
- Neri, F.; Dettori, D.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Maldotti, M.; Parlato, C.; Paliogiannis, P.; Oliviero, S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 2015, 34, 4168–4176. [Google Scholar] [CrossRef] [PubMed]
- Kai, M.; Niinuma, T.; Kitajima, H.; Yamamoto, E.; Harada, T.; Aoki, H.; Maruyama, R.; Toyota, M.; Sasaki, Y.; Sugai, T.; et al. TET1 Depletion Induces Aberrant CpG Methylation in Colorectal Cancer Cells. PLoS ONE 2016, 11, e0168281. [Google Scholar] [CrossRef]
- Kudo, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Asaoka, Y.; Ijichi, H.; Nagae, G.; Yoshida, H.; Aburatani, H.; Koike, K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012, 103, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2012, 13, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ficz, G.; Gribben, J.G. Loss of 5-hydroxymethylcytosine in cancer: Cause or consequence? Genomics 2014, 104, 352–357. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: Molecular mechanisms and therapeutic targets. Signal Transduct. Target Ther. 2023, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Tien, F.M.; Lu, H.H.; Lin, S.Y.; Tsai, H.C. Epigenetic remodeling of the immune landscape in cancer: Therapeutic hurdles and opportunities. J. Biomed. Sci. 2023, 30, 3. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Wang, A.; Yuan, Y.; Zhu, B.; Long, H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp. Hematol. Oncol. 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, Z.; Cheng, L. Tumor microenvironment heterogeneity an important mediator of prostate cancer progression and therapeutic resistance. NPJ Precis. Oncol. 2022, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kawakami, H. Mitochondrial Dysfunction and Nanocarrier-Based Treatments in Chronic Obstructive Pulmonary Disease (COPD). Oxygen 2023, 3, 394–406. [Google Scholar] [CrossRef]
- Dai, W.; Qiao, X.; Fang, Y.; Guo, R.; Bai, P.; Liu, S.; Li, T.; Jiang, Y.; Wei, S.; Na, Z.; et al. Epigenetics-targeted drugs: Current paradigms and future challenges. Signal Transduct. Target. Ther. 2024, 9, 332. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitsuhashi, R.; Sato, K.; Kawakami, H. Novel Epigenetics Control (EpC) Nanocarrier for Cancer Therapy Through Dual-Targeting Approach to DNA Methyltransferase and Ten-Eleven Translocation Enzymes. Epigenomes 2025, 9, 6. https://doi.org/10.3390/epigenomes9010006
Mitsuhashi R, Sato K, Kawakami H. Novel Epigenetics Control (EpC) Nanocarrier for Cancer Therapy Through Dual-Targeting Approach to DNA Methyltransferase and Ten-Eleven Translocation Enzymes. Epigenomes. 2025; 9(1):6. https://doi.org/10.3390/epigenomes9010006
Chicago/Turabian StyleMitsuhashi, Risa, Kiyoshi Sato, and Hiroyoshi Kawakami. 2025. "Novel Epigenetics Control (EpC) Nanocarrier for Cancer Therapy Through Dual-Targeting Approach to DNA Methyltransferase and Ten-Eleven Translocation Enzymes" Epigenomes 9, no. 1: 6. https://doi.org/10.3390/epigenomes9010006
APA StyleMitsuhashi, R., Sato, K., & Kawakami, H. (2025). Novel Epigenetics Control (EpC) Nanocarrier for Cancer Therapy Through Dual-Targeting Approach to DNA Methyltransferase and Ten-Eleven Translocation Enzymes. Epigenomes, 9(1), 6. https://doi.org/10.3390/epigenomes9010006