
Unveiling Gene Interactions in Alzheimer’s Disease by Integrating Genetic and Epigenetic Data with a Network-Based Approach

 ,
,
Abstract
1. Introduction
2. Results
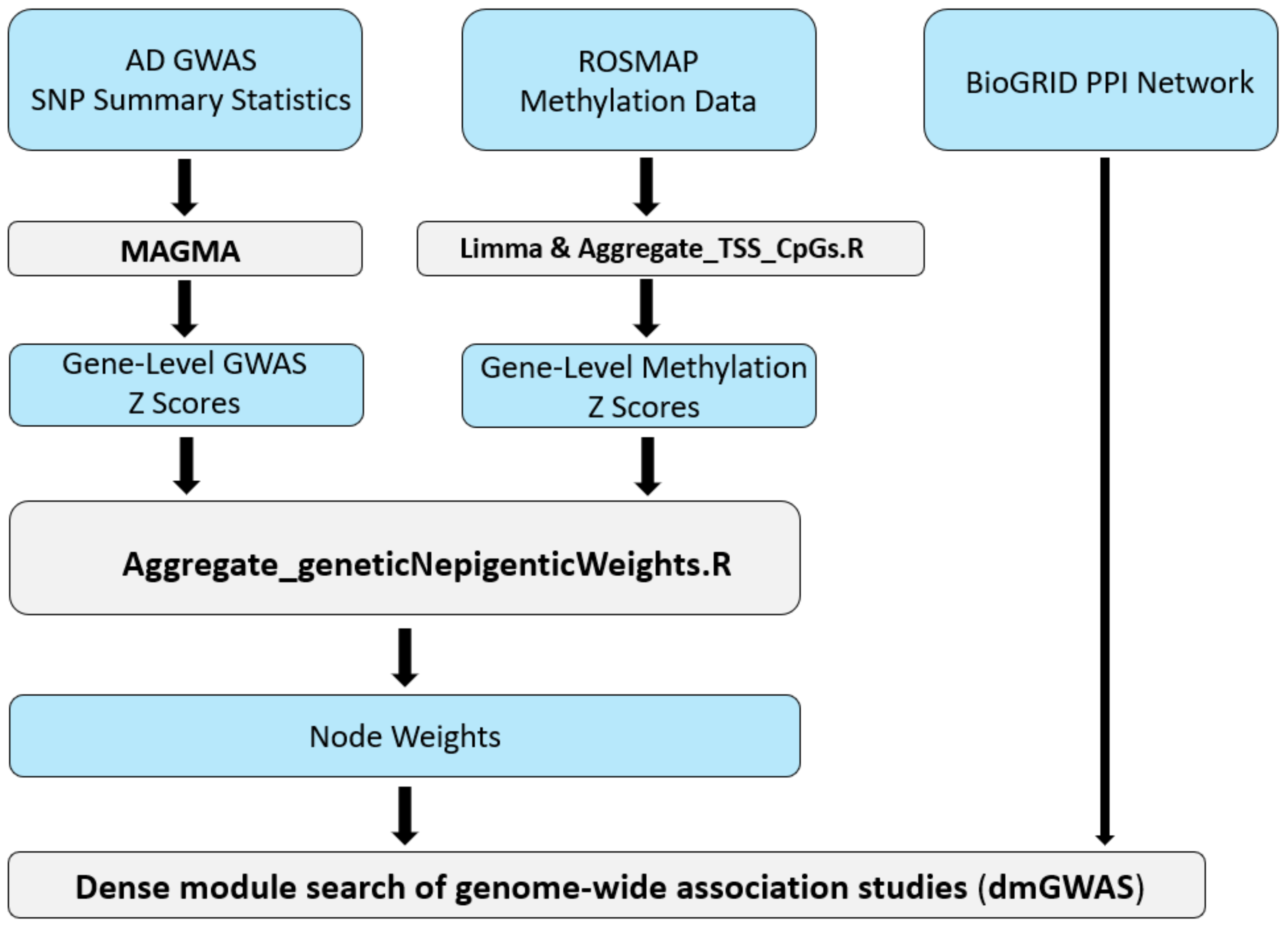
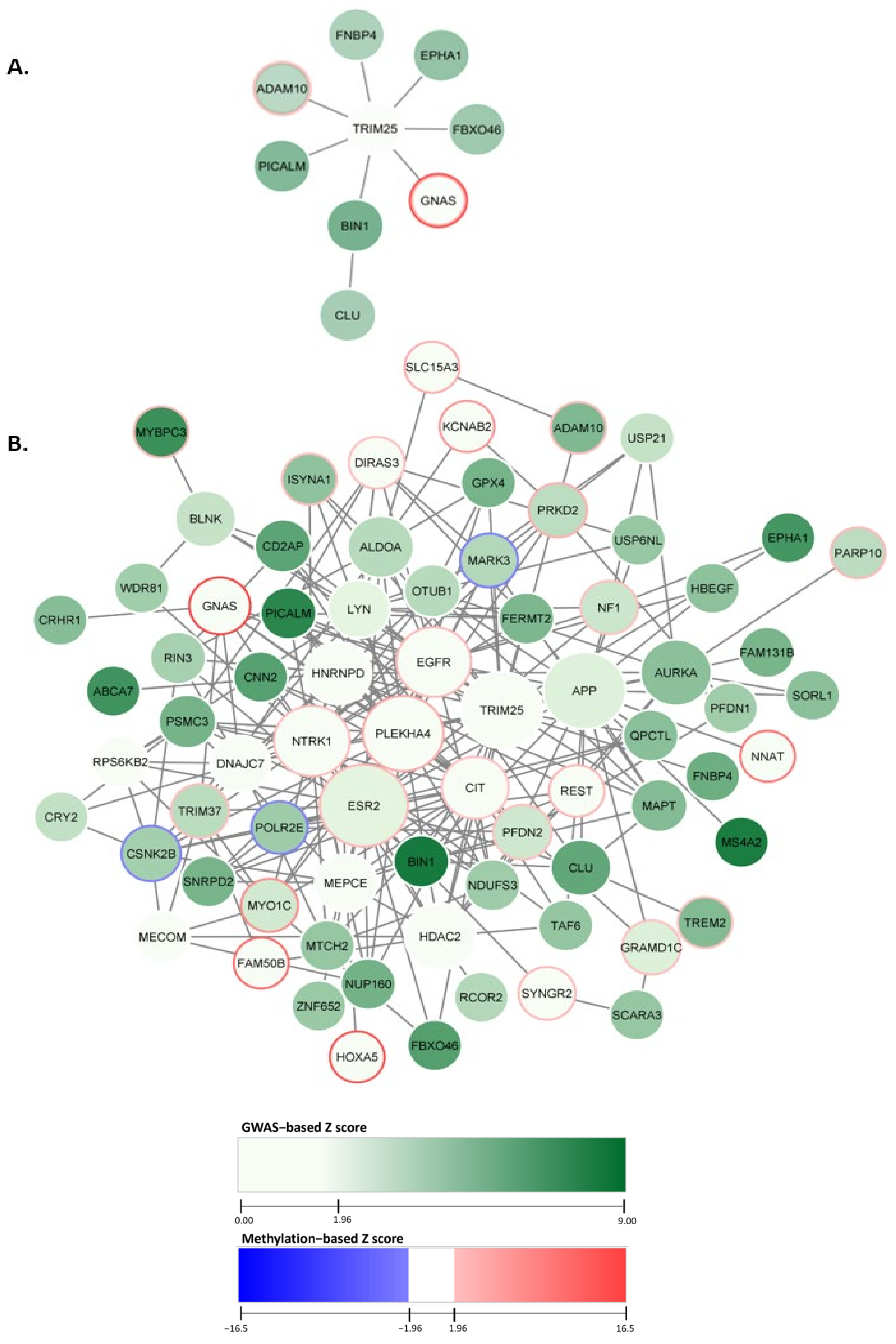
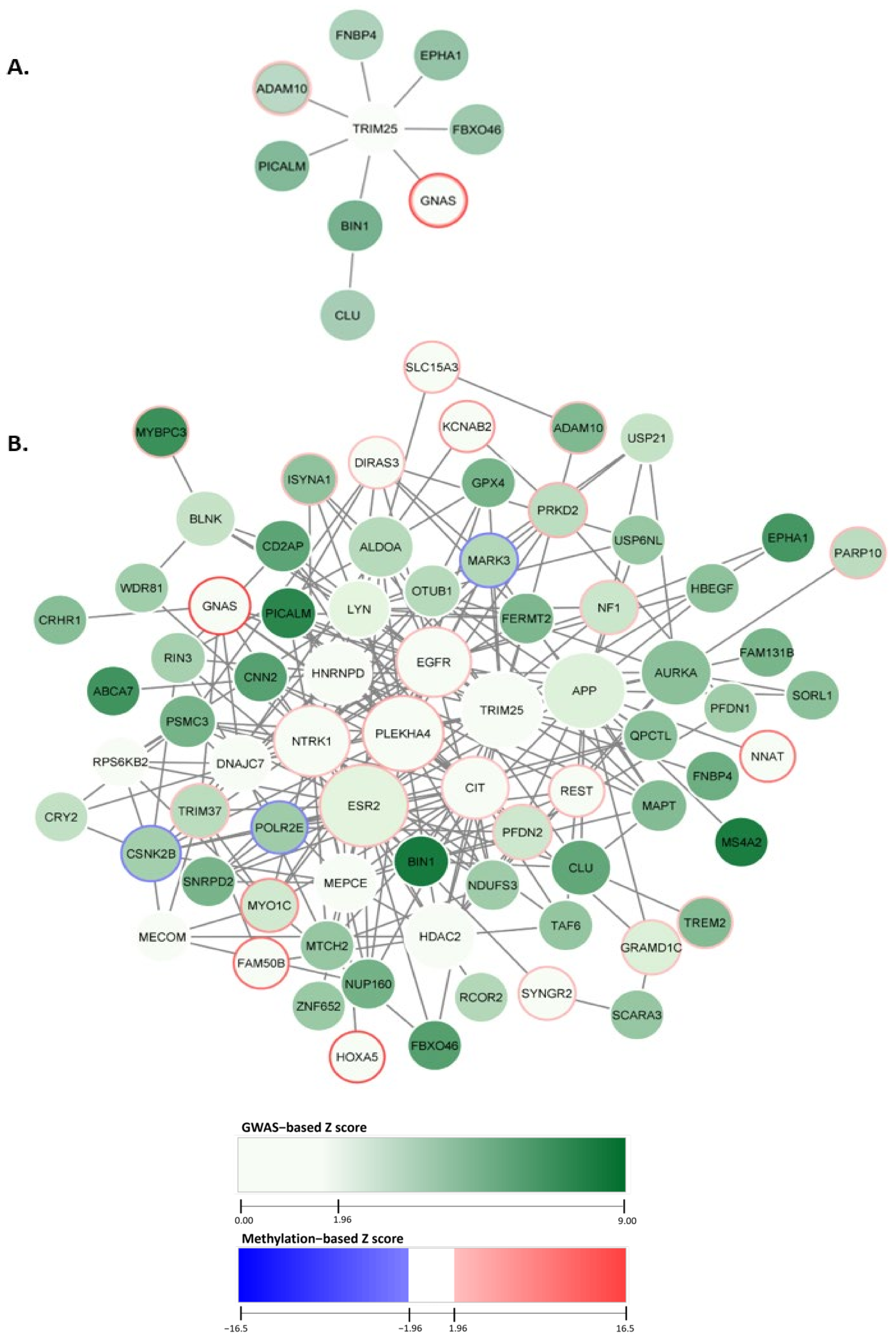
2.1. Constructing the AD-Associated Top Network Modules
2.2. AD-Associated Top Modules from dmGWAS Analysis
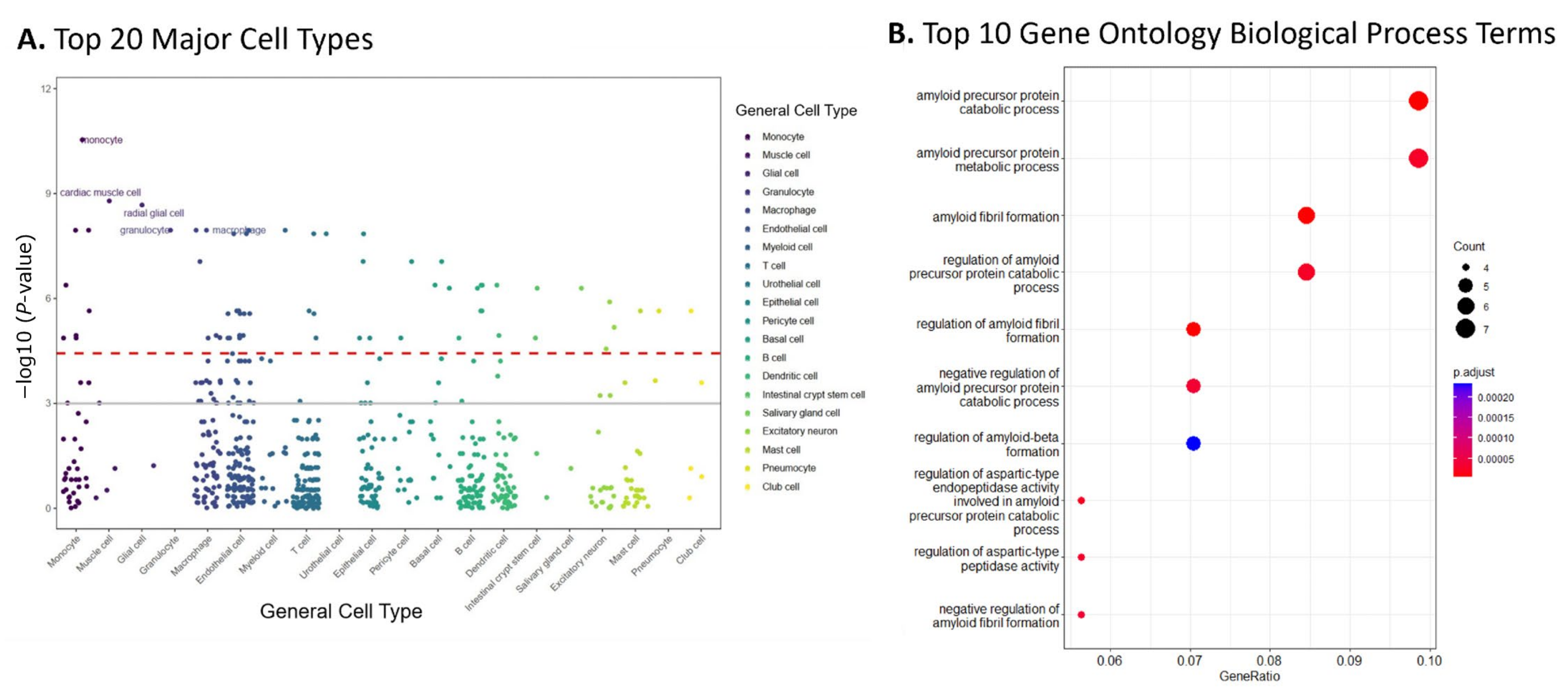
2.3. Enrichment Analyses of Top 10% Modules Suggested AD-Related Cell Type and Pathways
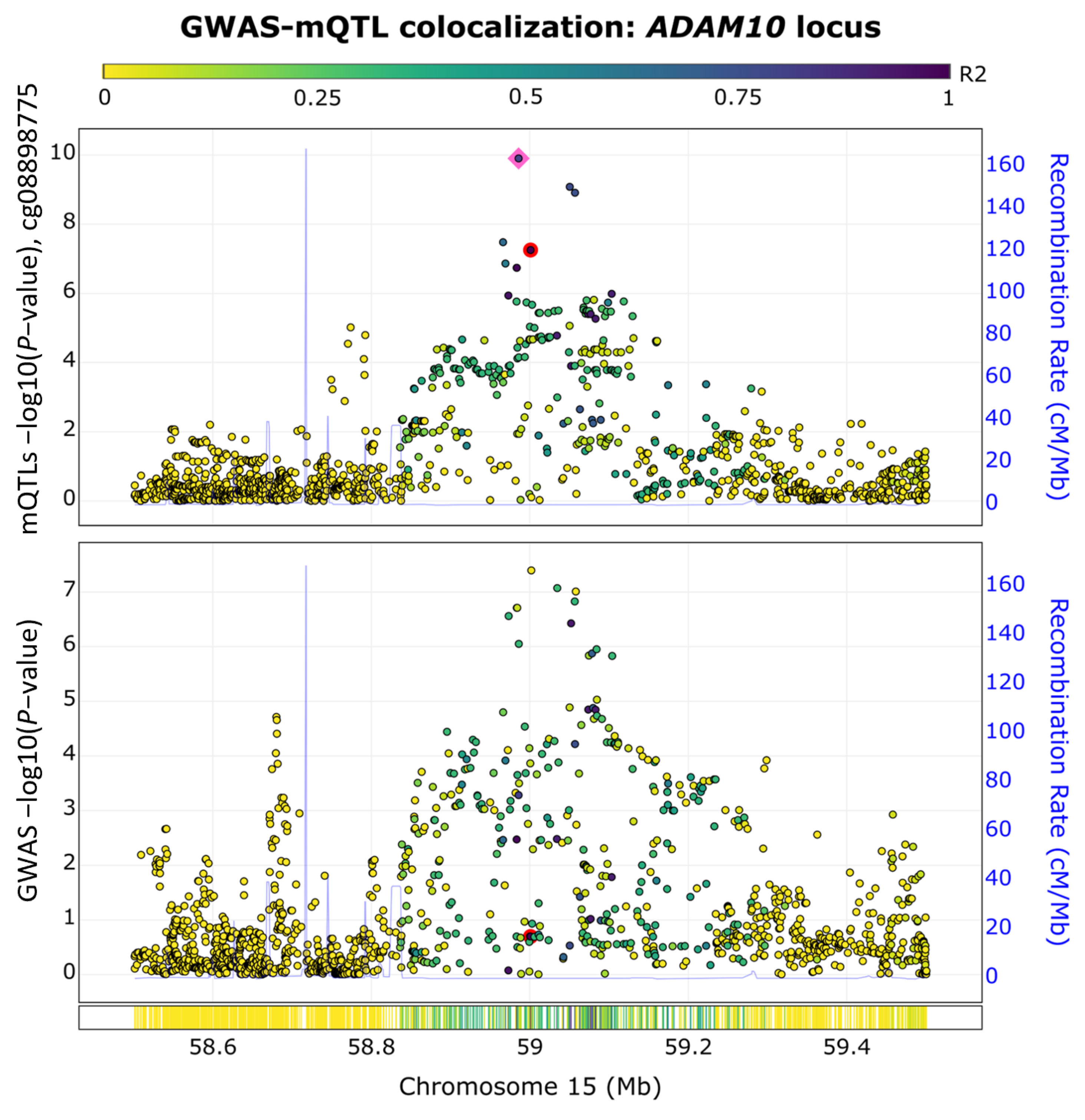
2.4. ADAM10 Exhibits Colocalized GWAS-Methylation Quantitative Trait Loci (QTL) Signals
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Collection of AD-Specific Multi-Omics Data
5.2. Pre-Processing of GWAS and DNA Methylation Data
5.3. Integration of GWAS and DNA Methylation Data Utilizing dmGWAS
5.4. Dense Module Searching Genome-Wide Association Studies Signals via dmGWAS
5.5. Cell Type and Gene Set Enrichment Analyses
5.6. Drug Target Analysis
5.7. GWAS-mQTL Colocalization Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Fiest, K.M.; Roberts, J.I.; Maxwell, C.J.; Hogan, D.B.; Smith, E.E.; Frolkis, A.; Cohen, A.; Kirk, A.; Pearson, D.; Pringsheim, T.; et al. The Prevalence and Incidence of Dementia Due to Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Can. J. Neurol. Sci. 2016, 43, S51. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018; Available online: https://www.alzint.org/u/WorldAlzheimerReport2018.pdf (accessed on 29 February 2024).
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the global burden of disease study 2016. Lancet 2021, 397, 1577. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, A.G. Genetics of complex diseases. J. Zhejiang Univ. B 2006, 7, 167–168. [Google Scholar]
- Iacono, D.; Zandi, P.; Gross, M.; Markesbery, W.R.; Pletnikova, O.; Rudow, G.; Troncoso, J.C. APOε2 and education in cognitively normal older subjects with high levels of AD pathology at autopsy: Findings from the nun study. Oncotarget 2015, 6, 14082. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452. [Google Scholar]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed alzheimer’s disease identifies new risk loci and implicates aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2020, 51, 404. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Cooper, S.; Liu, J.Z.; Barrio-Hernandez, I.; Bello, E.; Kumasaka, N.; Young, A.M.H.; Franklin, R.J.M.; Johnson, T.; Estrada, K.; et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 2021, 53, 392. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412. [Google Scholar] [CrossRef]
- Sanchez-Mut, J.; Graeff, J. Epigenetic alterations in Alzheimer’s disease. Front. Behav. Neurosci. 2015, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Han, L. CpG islands: Algorithms and applications in methylation studies. Biochem. Biophys. Res. Commun. 2010, 382, 643. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jia, P.; Zhao, Z. Deep4mC: Systematic assessment and computational prediction for DNA N4-methylcytosine sites by deep learning. Brief. Bioinform. 2020, 22, bbaa099. [Google Scholar] [CrossRef] [PubMed]
- Noroozi, R.; Ghafouri-Fard, S.; Pisarek, A.; Rudnicka, J.; Spólnicka, M.; Branicki, W.; Taheri, M.; Pośpiech, E. DNA methylation-based age clocks: From age prediction to age reversion. Ageing Res. Rev. 2021, 68, 101314. [Google Scholar] [CrossRef]
- Kabacik, S.; Lowe, D.; Fransen, L.; Leonard, M.; Ang, S.; Whiteman, C.; Corsi, S.; Cohen, H.; Felton, S.; Bali, R.; et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat. Aging 2022, 2, 484. [Google Scholar] [CrossRef]
- Hernando-Herraez, I.; Evano, B.; Stubbs, T.; Commere, P.; Jan Bonder, M.; Clark, S.; Andrews, S.; Tajbakhsh, S.; Reik, W. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat. Commun. 2019, 10, 4361. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic Predictor of Age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef]
- Jia, P.; Zheng, S.; Long, J.; Zheng, W.; Zhao, Z. dmGWAS: Dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics 2011, 27, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Manuel, A.M.; Dai, Y.; Jia, P.; Freeman, L.A.; Zhao, Z. A gene regulatory network approach harmonizes genetic and epigenetic signals and reveals repurposable drug candidates for multiple sclerosis. Hum. Mol. Genet. 2023, 32, 998. [Google Scholar] [CrossRef] [PubMed]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Børte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for alzheimer’s disease. Nat. Genet. 2021, 53, 1276. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Wilson, R.S. Overview and findings from the religious orders study. Curr. Alzheimer Res. 2012, 9, 628–645. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Stark, C.; Breitkreutz, B.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Hu, R.; Liu, A.; Cho, K.S.; Manuel, A.M.; Li, X.; Dong, X.; Jia, P.; Zhao, Z. WebCSEA: Web-based cell-type-specific enrichment analysis of genes. Nucleic Acids Res. 2022, 50, W782–W790. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2021, 50, D1398–D1407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Klein, A.; Sang, J.; Choi, J.; Brown, K.M. ezQTL: A Web Platform for Interactive Visualization and Colocalization of QTLs and GWAS Loci. Genom. Proteom. Bioinform. 2022, 20, 541. [Google Scholar] [CrossRef] [PubMed]
- Mcrae, A.F.; Marioni, R.E.; Shah, S.; Yang, J.; Powell, J.E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Bowdler, L.; Painter, J.N.; et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci. Rep. 2018, 8, 17605. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; Van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Nebel, A.; Kleindorp, R.; Caliebe, A.; Nothnagel, M.; Blanché, H.; Junge, O.; Wittig, M.; Ellinghaus, D.; Flachsbart, F.; Wichmann, H.; et al. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech. Ageing Dev. 2011, 132, 324. [Google Scholar] [CrossRef] [PubMed]
- Kodam, P.; Sai Swaroop, R.; Pradhan, S.S.; Sivaramakrishnan, V.; Vadrevu, R. Integrated multi-omics analysis of Alzheimer’s disease shows molecular signatures associated with disease progression and potential therapeutic targets. Sci. Rep. 2023, 13, 3695. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.M.; Curtis, M.A.; Dragunow, M. Global changes in DNA methylation and hydroxymethylation in alzheimer’s disease human brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2016, 77, 43. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Dowzell, K.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carrasquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Prokic, I.; Cowling, B.S.; Laporte, J. Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. 2014, 92, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.G. Endocytosis. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 197–199. [Google Scholar]
- Heckmann, B.L.; Green, D.R. Endocytosis in β-amyloid biology and Alzheimer’s disease. In Autophagy Dysfunction in Alzheimer’s Disease and Dementia; Academic Press: Cambridge, MA, USA, 2022; p. 111. [Google Scholar]
- Turan, S.; Bastepe, M. GNAS spectrum of disorders. Curr. Osteoporos. Rep. 2015, 13, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, K.; Jeon, Y.; Oh, J.-H.; Jeong, S.-Y.; Song, I.-S.; Lee, D.-S.; Kim, N.-S. Identification of genes related to Parkinson’s disease using expressed sequence tags. DNA Res. 2006, 13, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Martín-Vicente, M.; Medrano, L.M.; Resino, S.; García-Sastre, A.; Martínez, I. TRIM25 in the regulation of the antiviral innate immunity. Front. Immunol. 2017, 8, 1187. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Levine, H. Alzheimer’s Disease and the β-Amyloid Peptide. J. Alzheimer’s Dis. 2010, 19, 311. [Google Scholar] [CrossRef]
- Peron, R.; Vatanabe, I.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, E.; Schmid, J.P.; Jurinovic, V.; Becker, M.; Wirth, A.; Ludwig, R.; Kreissig, S.; Angel, T.V.D.; Amend, D.; Hunt, K.; et al. Combined proteomics and CRISPR-Cas9 screens in PDX identify ADAM10 as essential for leukemia in vivo. Mol. Cancer 2023, 22, 107. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533. [Google Scholar] [CrossRef] [PubMed]
- Drakulic, D.; Schwirtlich, M.; Petrovic, I.; Mojsin, M.; Milivojevic, M.; Kovacevic-Grujicic, N.; Stevanovic, M. Current Opportunities for Targeting Dysregulated Neurodevelopmental Signaling Pathways in Glioblastoma. Cells 2022, 11, 2530. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Suh, J.; Romano, D.; Truong, M.H.; Mullin, K.; Hooli, B.; Norton, D.; Tesco, G.; Elliott, K.; Wagner, S.L.; et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum. Mol. Genet. 2024, 18, 3987. [Google Scholar]
- Huang, W.; Chen, W.; Jiang, L.; Yang, Y.; Yao, L.; Li, K. Influence of ADAM10 Polymorphisms on Plasma Level of Soluble Receptor for Advanced Glycation End Products and The Association with Alzheimer’s Disease Risk. Front. Genet. 2018, 9, 540. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xie, Y.; Meng, X.; Kang, J. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, N.; Wei, W.; Zhang, Z.; Li, H. Integrated analysis of weighted gene coexpression network analysis identifying six genes as novel biomarkers for Alzheimer’s disease. Oxidative Med. Cell. Longev. 2021, 2021, 9918498. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.K.; Lee, Y.R.; Ong, L.; Gold, M.; Kalali, A.; Sarkar, J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J. Alzheimer’s Dis. 2022, 87, 83. [Google Scholar] [CrossRef] [PubMed]
- Doraiswamy, P.M.; Sperling, R.A.; Coleman, R.E.; Johnson, K.A.; Reiman, E.M.; Davis, M.D.; Grundman, M.; Sabbagh, M.N.; Sadowsky, C.H.; Fleisher, A.S.; et al. Amyloid- assessed by florbetapir F 18 PET and 18-month cognitive decline: A multicenter study. Neurology 2012, 79, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Futch, H.S.; Croft, C.L.; Truong, V.Q.; Krause, E.G.; Golde, T.E. Targeting psychologic stress signaling pathways in Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Karlowsky, J.A.; Nichol, K.; Zhanel, G.G. Telavancin: Mechanisms of Action, In Vitro Activity, and Mechanisms of Resistance. Clin. Infect. Dis. 2015, 61, S58–S68. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2017, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Colclough, N.; Chen, K.; Johnström, P.; Strittmatter, N.; Yan, Y.; Wrigley, G.L.; Schou, M.; Goodwin, R.; Varnäs, K.; Adua, S.J.; et al. Preclinical Comparison of the Blood–brain barrier Permeability of Osimertinib with Other EGFR TKIs. Clin. Cancer Res. 2023, 27, 189. [Google Scholar] [CrossRef] [PubMed]
- Advani, D.; Kumar, P. Therapeutic Targeting of Repurposed Anticancer Drugs in Alzheimer’s Disease: Using the Multiomics Approach. Am. Chem. Soc. Omega 2021, 6, 13870–13887. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lee, J.H.; Pang, D.; Temkin, A.; Park, N.; Janicki, S.C.; Zigman, W.B.; Silverman, W.; Tycko, B.; Schupf, N. Estrogen receptor-beta variants are associated with increased risk of alzheimer’s disease in women with down syndrome. Dement. Geriatr. Cogn. Disord. 2011, 32, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.N.M.; Hornberger, M.; Ritchie, C.W.; Minihane, A.M. Hormone replacement therapy is associated with improved cognition and larger brain volumes in at-risk APOE4 women: Results from the European Prevention of Alzheimer’s Disease (EPAD) cohort. Alzheimer’s Res. Ther. 2023, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wu, J.; Li, S.; Wang, S.; Zhang, J.; Wang, Y.; Yan, Y.; Hu, H.; Xiong, M.; Bai, C. NTRK1 knockdown induces mouse cognitive impairment and hippocampal neuronal damage through mitophagy suppression via inactivating the AMPK/ULK1/FUNDC1 pathway. Cell Death Discov. 2023, 9, 404. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, C.; Zhao, Z.; Lu, D.; Zheng, G. Uncovering neuroinflammation-related modules and potential repurposing drugs for Alzheimer’s disease through multi-omics data integrative analysis. Front. Aging Neurosci. 2023, 15, 1161405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Zhao, D.; Yu, X.; Shen, X.; Zhou, Y.; Fernandes, B.S.; Enduru, N.; Jia, P.; Zhao, Z. TTD: Therapeutic Target Database describing target druggability information. 2023, 52, D1465.
- Wang, Q.; Yu, H.; Zhao, Z.; Jia, P. EW_dmGWAS: Edge-weighted dense module search for genome-wide association studies and gene expression profiles. Bioinformatics 2015, 31, 2591. [Google Scholar] [CrossRef]
- Liu, A.; Manuel, A.M.; Dai, Y.; Fernandes, B.S.; Enduru, N.; Jia, P.; Zhao, Z. Identifying candidate genes and drug targets for Alzheimer’s disease by an integrative network approach using genetic and brain region-specific proteomic data. Hum. Mol. Genet. 2022, 31, 3341. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 20 December 2022).
- Ware, E.B.; Faul, J.D.; Mitchell, C.M.; Bakulski, K.M. Considering the APOE locus in alzheimer’s disease polygenic scores in the health and retirement study: A longitudinal panel study. BMC Med. Genom. 2020, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Kim, S.C.; Lee, S.J.; Lee, W.J.; Yum, Y.N.; Kim, J.H.; Sohn, S.; Park, J.H.; Lee, J.; Lim, J.; Kwon, S.W. Stouffer’s Test in a Large Scale Simultaneous Hypothesis Testing. PLoS ONE 2013, 8, e63290. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Data Type | Sample Size |
|---|---|---|
| Wightman et al. | GWAS | N = 398,058 (Case = 39,918, Control = 358,140) |
| ROSMAP | DNA methylation | N = 549 (Case = 325, Control = 224) |
| BioGRID: reference network | Protein–protein interaction | N = 536,844 edges M = 19,087 proteins |
| Target Gene | Target Name | Drug Compound | Indication |
|---|---|---|---|
| APP | Amyloid beta A4 protein | Florbetapir F-18 | Diagnostic Imaging [ICD-11: N.A.]] |
| CRHR1 | Corticotropin-releasing factor receptor 1 | Telavancin | Staphylococcus infection [ICD-11: 1B5Y] |
| EGFR | Epidermal growth factor receptor | Osimertinib | Non-small-cell lung cancer [ICD-11: 2C25.Y, ICD-9: 162] |
| ESR2 | Estrogen receptor beta | Conjugated Estrogens | Menopause symptom [ICD-11: GA30.0, ICD-9: 627.2] Vasomotor symptom [ICD-11: CA08, ICD-10: J30-J39, J30, ICD-9: 627.2] |
| NTRK1 | Tropomyosin-related kinase A | Entrectinib | Non-small cell lung cancer [ICD-11: 2C25] Neuroblastoma [ICD-11: 2D11.2] (Phase 1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanders, K.L.; Manuel, A.M.; Liu, A.; Leng, B.; Chen, X.; Zhao, Z. Unveiling Gene Interactions in Alzheimer’s Disease by Integrating Genetic and Epigenetic Data with a Network-Based Approach. Epigenomes 2024, 8, 14. https://doi.org/10.3390/epigenomes8020014
Sanders KL, Manuel AM, Liu A, Leng B, Chen X, Zhao Z. Unveiling Gene Interactions in Alzheimer’s Disease by Integrating Genetic and Epigenetic Data with a Network-Based Approach. Epigenomes. 2024; 8(2):14. https://doi.org/10.3390/epigenomes8020014
Chicago/Turabian StyleSanders, Keith L., Astrid M. Manuel, Andi Liu, Boyan Leng, Xiangning Chen, and Zhongming Zhao. 2024. "Unveiling Gene Interactions in Alzheimer’s Disease by Integrating Genetic and Epigenetic Data with a Network-Based Approach" Epigenomes 8, no. 2: 14. https://doi.org/10.3390/epigenomes8020014
APA StyleSanders, K. L., Manuel, A. M., Liu, A., Leng, B., Chen, X., & Zhao, Z. (2024). Unveiling Gene Interactions in Alzheimer’s Disease by Integrating Genetic and Epigenetic Data with a Network-Based Approach. Epigenomes, 8(2), 14. https://doi.org/10.3390/epigenomes8020014