
Angio-Long Noncoding RNA MALAT1 (rs3200401) and MIAT (rs1061540) Gene Variants in Ovarian Cancer

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Characteristics of the Study Population
2.2. Pathological and Molecular Assessment
2.3. Subgroup Analysis of Malignant Epithelial Tumors
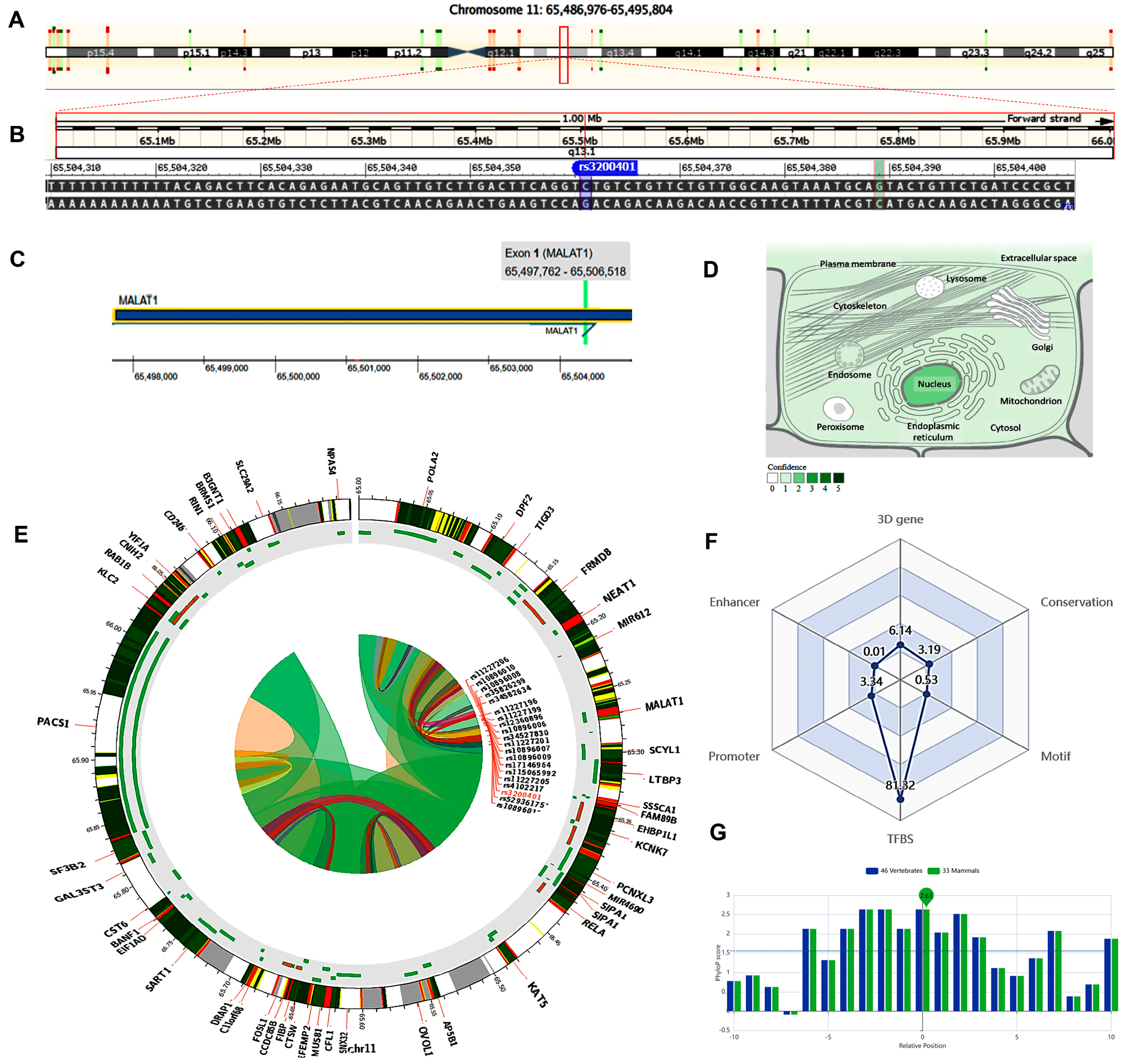
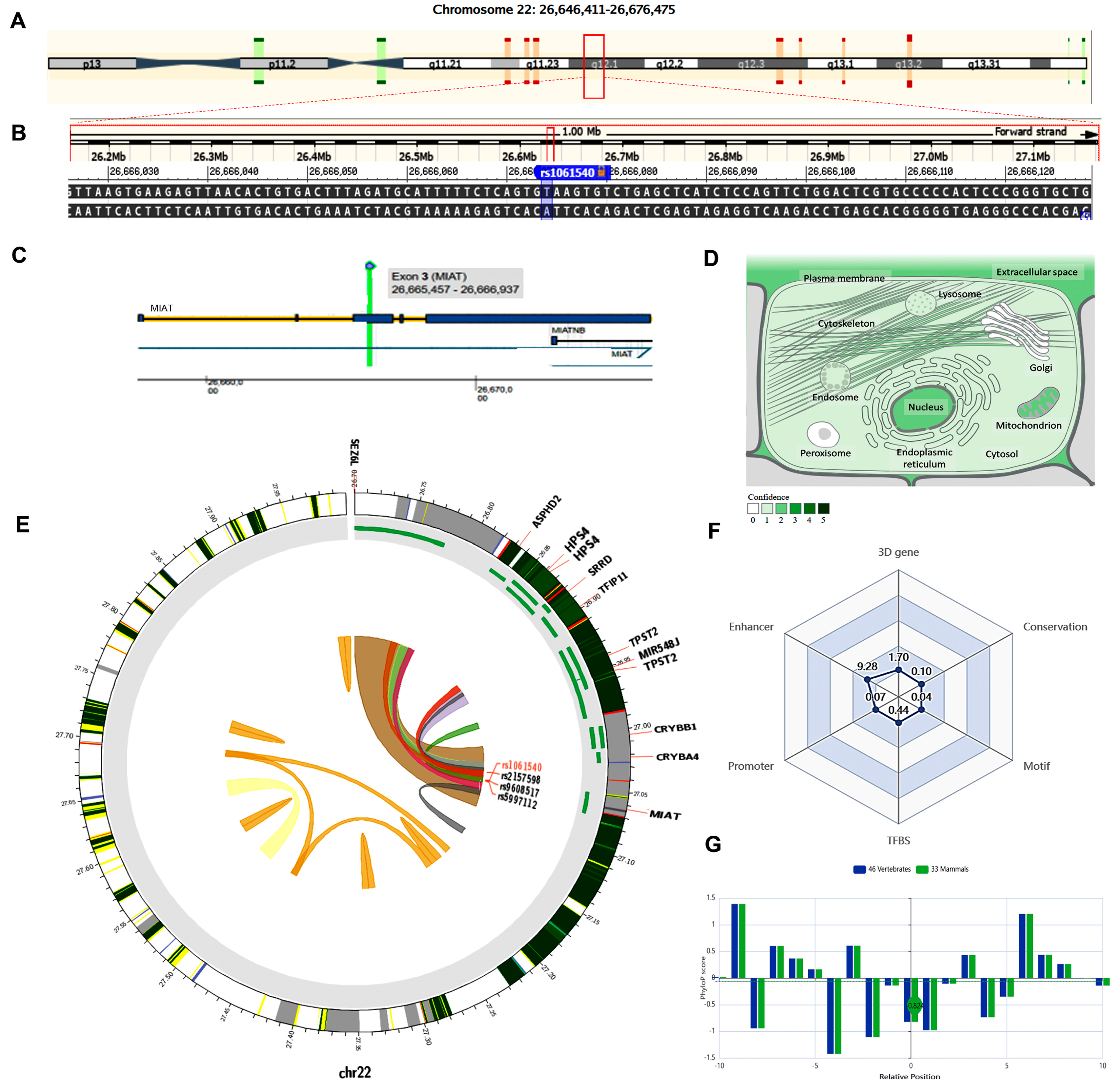
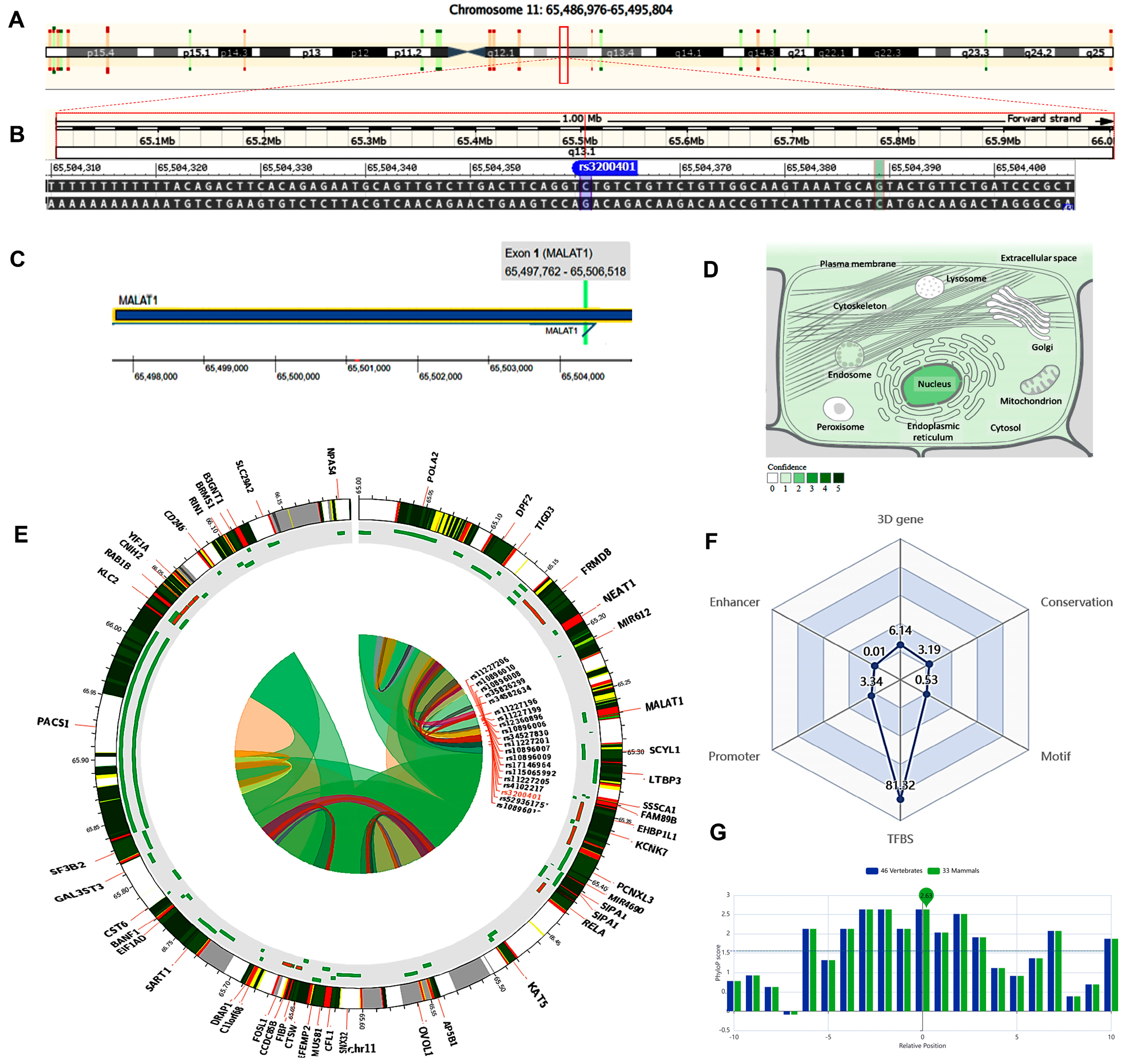
2.4. In Silico Data Analysis and Variant Functional Annotation
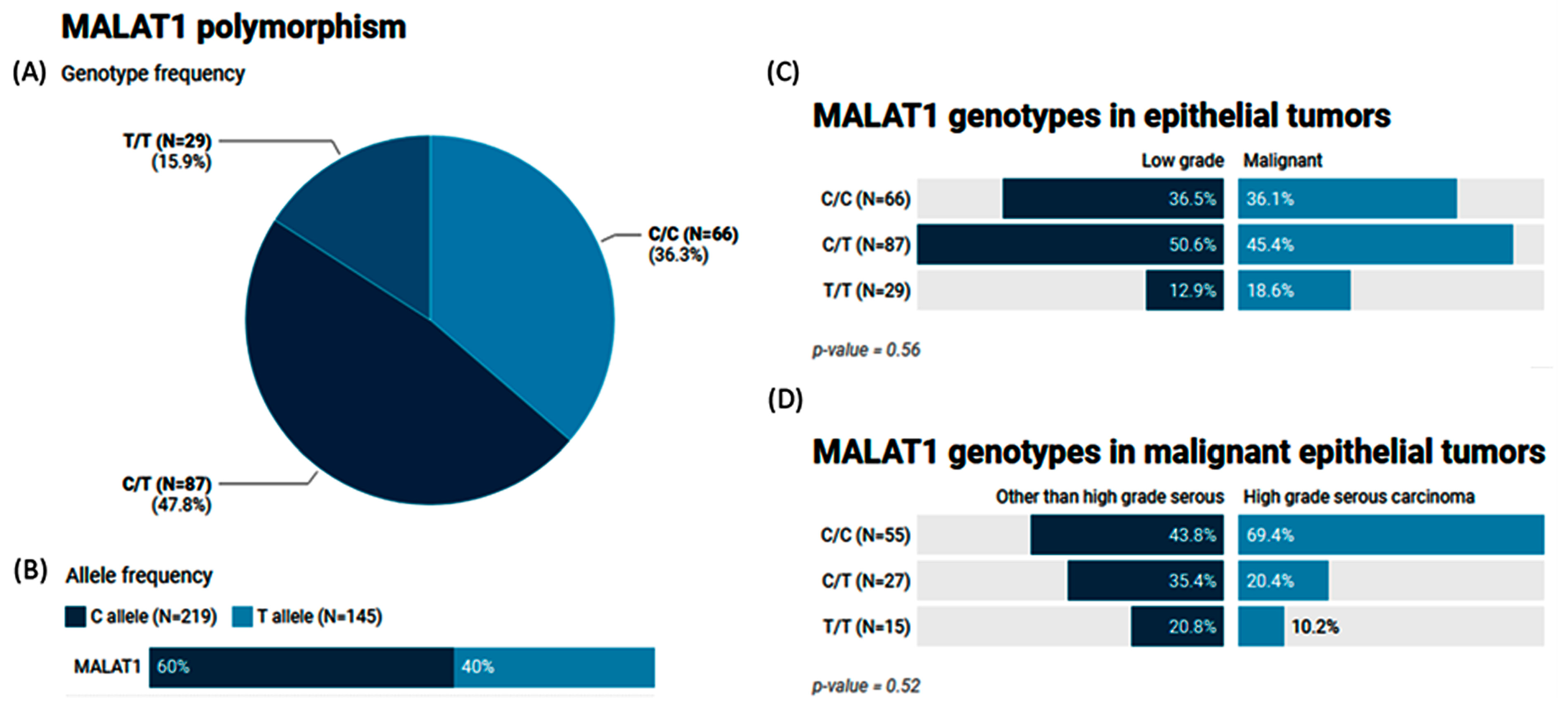
2.5. MALAT1 (rs3200401) Variant Genotype and Allele Frequencies in Patients with Ovarian Tumors
2.6. The Impact of the MALAT1 (rs3200401) Variant on Ovarian Cancer Risk
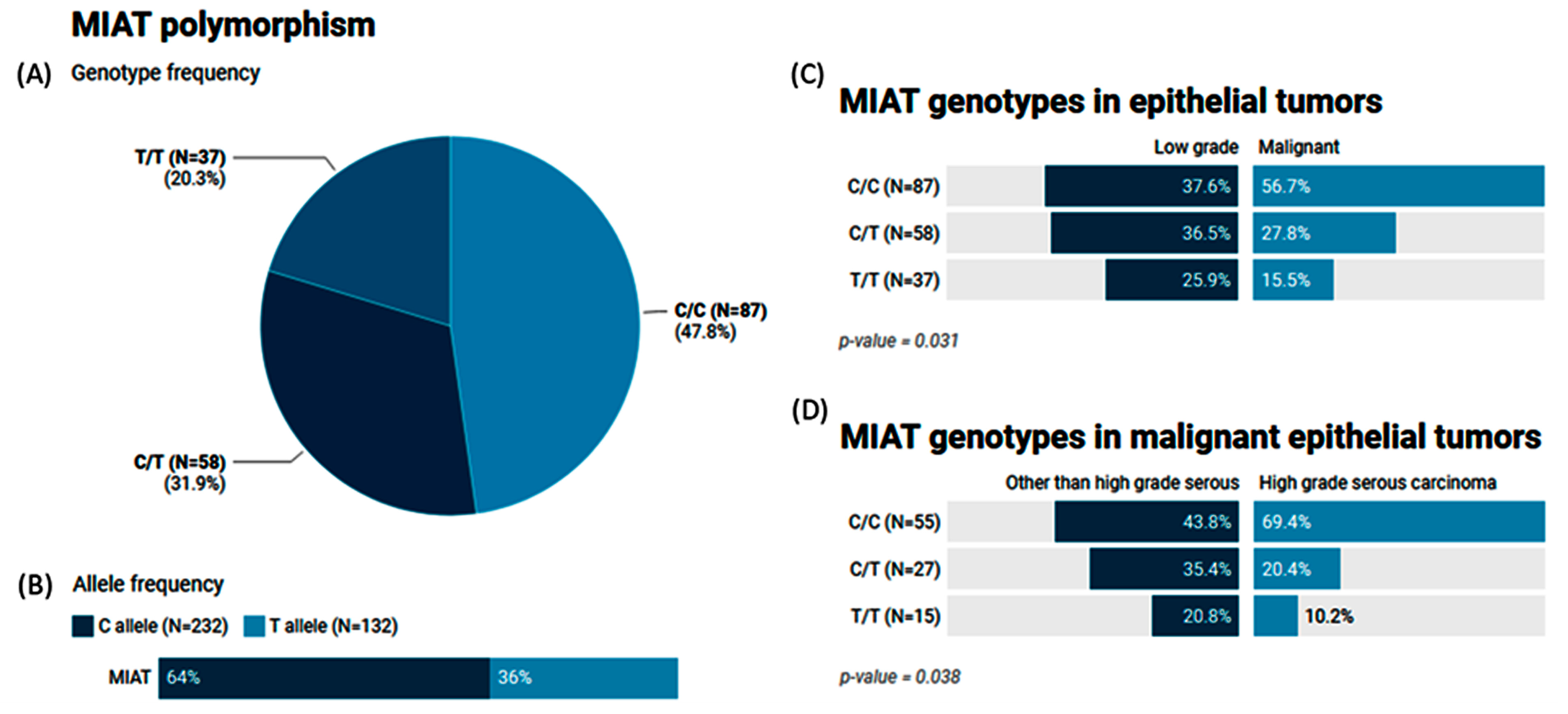
2.7. Genotype and Allele Frequencies of the MIAT (rs1061540) Variant in Patients with Ovarian Tumors
2.8. The Impact of the MIAT (rs1061540) Variant on Ovarian Cancer Risk
2.9. Genotype Combination and Ovarian Cancer Risk
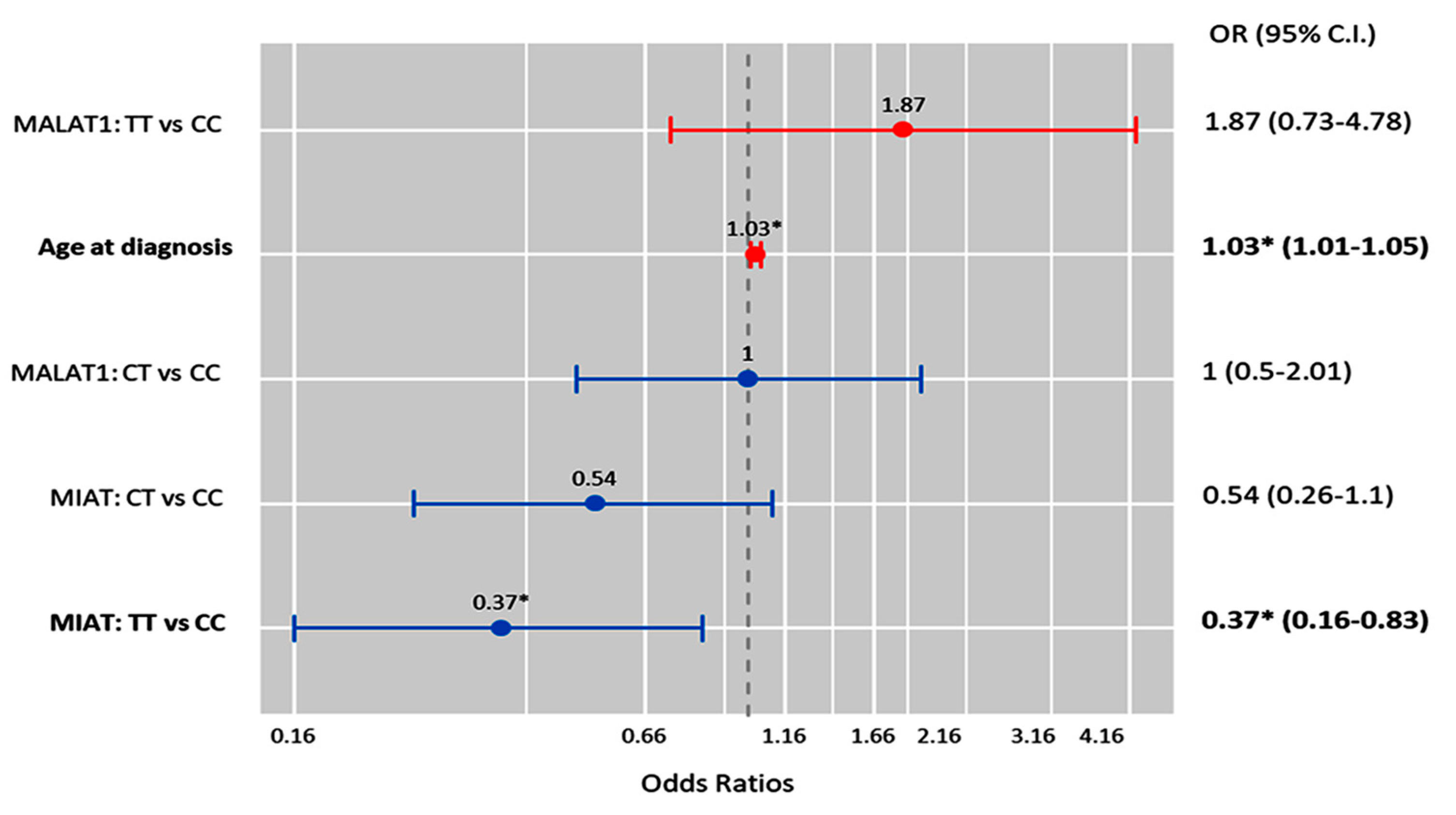
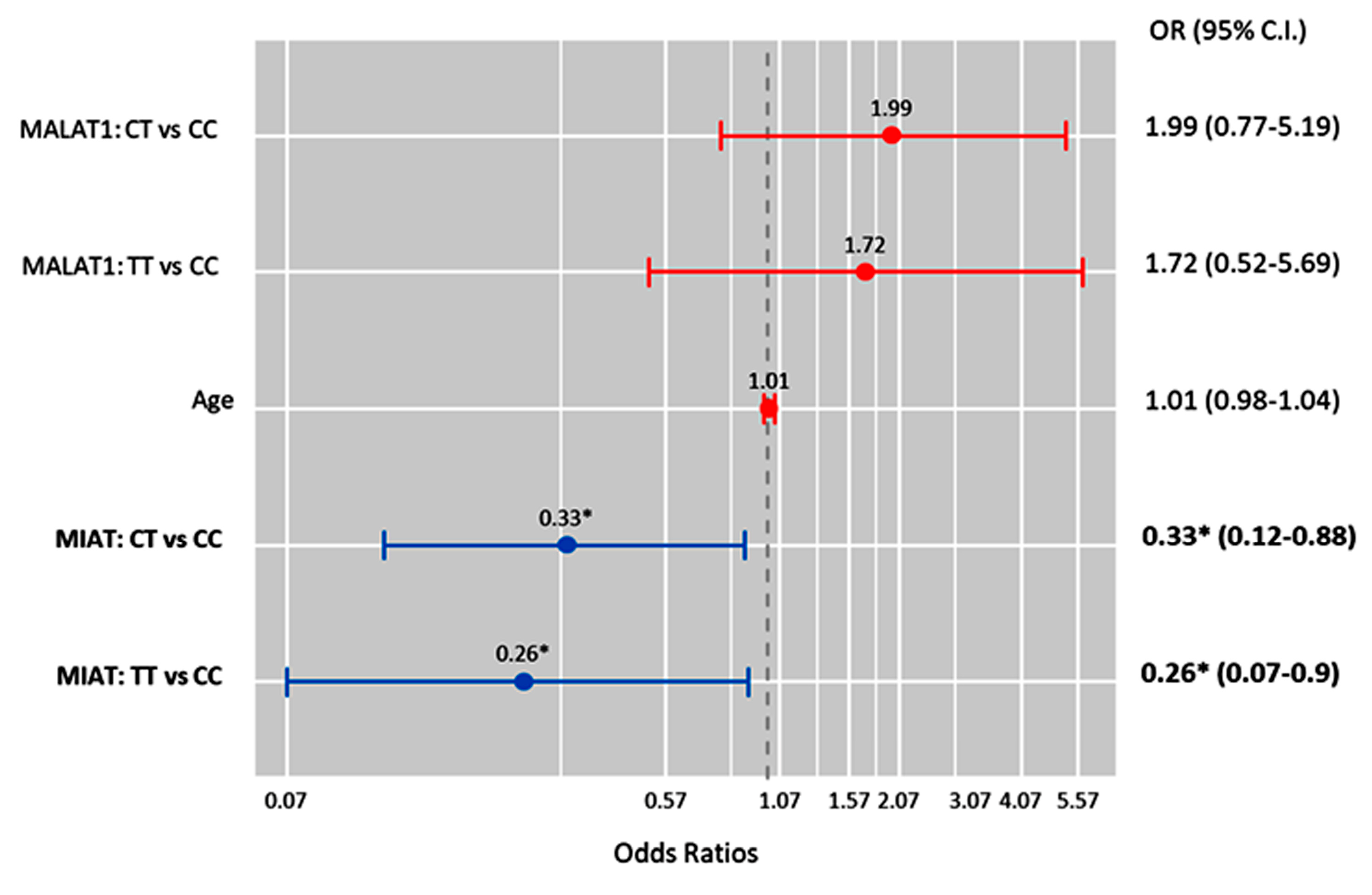
2.10. Multivariate Cox Regression Analysis
3. Discussion
4. Materials and Methods
4.1. Archived Tissue Sampling
4.2. Pathological Assessment
4.3. Criteria for Selecting the lncRNA SNPs and In Silico Data Analysis
4.4. Analysis of LncRNA Gene Variants MALAT1 rs3200401 and MIAT rs1061540
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef]
- Coburn, S.B.; Bray, F.; Sherman, M.E.; Trabert, B. International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int. J. Cancer 2017, 140, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Mazidimoradi, A.; Momenimovahed, Z.; Allahqoli, L.; Tiznobaik, A.; Hajinasab, N.; Salehiniya, H.; Alkatout, I. The global, regional and national epidemiology, incidence, mortality, and burden of ovarian cancer. Health Sci. Rep. 2022, 5, e936. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, R.; Talhouk, A.; Eshragh, S.; Lau, S.; Cheung, D.; Chow, C.; Le, N.; Cook, L.S.; Wilkinson, N.; McDermott, J.; et al. Morphologic and Molecular Characteristics of Mixed Epithelial Ovarian Cancers. Am. J. Surg. Pathol. 2015, 39, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yang, Y.; Zhou, N.; Tang, K.; Lau, W.B.; Lau, B.; Wang, W.; Xu, L.; Yang, Z.; Huang, S.; et al. Epigenetics in ovarian cancer: Premise, properties, and perspectives. Mol. Cancer 2018, 17, 109. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Gayther, S.A. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol. Oncol. 2009, 3, 138–150. [Google Scholar] [CrossRef]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 genes. BioMed Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell. Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Akrami, R.; Jacobsen, A.; Hoell, J.; Schultz, N.; Sander, C.; Larsson, E. Comprehensive analysis of long non-coding RNAs in ovarian cancer reveals global patterns and targeted DNA amplification. PLoS ONE 2013, 8, e80306. [Google Scholar] [CrossRef]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef]
- Worku, T.; Bhattarai, D.; Ayers, D.; Wang, K.; Wang, C.; Rehman, Z.U.; Talpur, H.S.; Yang, L. Long Non-Coding RNAs: The New Horizon of Gene Regulation in Ovarian Cancer. Cell. Physiol. Biochem. 2017, 44, 948–966. [Google Scholar] [CrossRef]
- Toraih, E.A.; Sedhom, J.A.; Haidari, M.; Fawzy, M.S. Applications of non-coding RNAs in renal cancer patients. In Clinical Applications of Non-Coding RNAs in Cancer; Elsevier: Amsterdam, The Netherlands, 2022; pp. 211–284. [Google Scholar]
- Abildgaard, C.; Do Canto, L.M.; Steffensen, K.D.; Rogatto, S.R. Long Non-coding RNAs Involved in Resistance to Chemotherapy in Ovarian Cancer. Front. Oncol. 2019, 9, 1549. [Google Scholar] [CrossRef]
- Mitra, R.; Chen, X.; Greenawalt, E.J.; Maulik, U.; Jiang, W.; Zhao, Z.; Eischen, C.M. Decoding critical long non-coding RNA in ovarian cancer epithelial-to-mesenchymal transition. Nat. Commun. 2017, 8, 1604. [Google Scholar] [CrossRef]
- Oncul, S.; Amero, P.; Rodriguez-Aguayo, C.; Calin, G.A.; Sood, A.K.; Lopez-Berestein, G. Long non-coding RNAs in ovarian cancer: Expression profile and functional spectrum. RNA Biol. 2020, 17, 1523–1534. [Google Scholar] [CrossRef]
- Lu, Y.M.; Guo, Y.R.; Zhou, M.Y.; Wang, Y. Expression and clinical significance of lncRNA BC041954 in ovarian cancer. Exp. Ther. Med. 2022, 23, 408. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Dostie, J. The Talented LncRNAs: Meshing into Transcriptional Regulatory Networks in Cancer. Cancers 2023, 15, 3433. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, G. LncRNAs: The good, the bad, and the unknown. Biochem. Cell. Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Ala, U. Competing Endogenous RNAs, Non-Coding RNAs and Diseases: An Intertwined Story. Cells 2020, 9, 1574. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.X.; Jiang, Y.P.; Tang, Y.L.; Liang, X.H. The crosstalk between lncRNA and microRNA in cancer metastasis: Orchestrating the epithelial-mesenchymal plasticity. Oncotarget 2017, 8, 12472–12483. [Google Scholar] [CrossRef] [PubMed]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
- Loizzi, V.; Del Vecchio, V.; Gargano, G.; De Liso, M.; Kardashi, A.; Naglieri, E.; Resta, L.; Cicinelli, E.; Cormio, G. Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer. Int. J. Mol. Sci. 2017, 18, 1967. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Wang, S. Angio-LncRs: LncRNAs that regulate angiogenesis and vascular disease. Theranostics 2018, 8, 3654–3675. [Google Scholar] [CrossRef] [PubMed]
- Michalik, K.M.; You, X.; Manavski, Y.; Doddaballapur, A.; Zörnig, M.; Braun, T.; John, D.; Ponomareva, Y.; Chen, W.; Uchida, S.; et al. Long non-coding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 2014, 114, 1389–1397. [Google Scholar] [CrossRef]
- Yan, B.; Yao, J.; Liu, J.Y.; Li, X.M.; Wang, X.Q.; Li, Y.J.; Tao, Z.F.; Song, Y.C.; Chen, Q.; Jiang, Q. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ. Res. 2015, 116, 1143–1156. [Google Scholar] [CrossRef]
- Minotti, L.; Agnoletto, C.; Baldassari, F.; Corrà, F.; Volinia, S. SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies? High-Throughput 2018, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhuang, S.; Hu, Y.; Xi, L.; Deng, L.; Sheng, H.; Shen, W. Associations between polymorphisms of long non-coding RNA MEG3 and risk of colorectal cancer in Chinese. Oncotarget 2016, 7, 19054–19059. [Google Scholar] [CrossRef]
- Guo, L.; Lu, X.; Zheng, L.; Liu, X.; Hu, M. Association of Long Non-Coding RNA HOTAIR Polymorphisms with Cervical Cancer Risk in a Chinese Population. PLoS ONE 2016, 11, e0160039. [Google Scholar] [CrossRef]
- Pan, W.; Wu, C.; Su, Z.; Duan, Z.; Li, L.; Mi, F.; Li, C. Genetic polymorphisms of non-coding RNAs associated with increased head and neck cancer susceptibility: A systematic review and meta-analysis. Oncotarget 2017, 8, 62508–62523. [Google Scholar] [CrossRef]
- Petkevicius, V.; Streleckiene, G.; Balciute, K.; Link, A.; Leja, M.; Malfertheiner, P.; Skieceviciene, J.; Kupcinskas, J. Association of Long Non-Coding RNA Polymorphisms with Gastric Cancer and Atrophic Gastritis. Genes 2020, 11, 1505. [Google Scholar] [CrossRef]
- Kattan, S.W.; Hobani, Y.H.; Shaheen, S.; Mokhtar, S.H.; Hussein, M.H.; Toraih, E.A.; Fawzy, M.S.; Abdalla, H.A. Association of cyclin-dependent kinase inhibitor 2B antisense RNA 1 gene expression and rs2383207 variant with breast cancer risk and survival. Cell. Mol. Biol. Lett. 2021, 26, 14. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.; Alshammari, E.M.; Mokhtar, S.H.; Alshanwani, A.R.; Toraih, E.A.; Ibrahiem, A.T.; Fawzy, M.S.; Maher, S.A. PUNISHER rs12318065 C>A transversion: A putative somatic driver mutation for poor prognosis in colon cancer. Biosci. Rep. 2022, 42, BSR20220465. [Google Scholar] [CrossRef]
- Hobani, Y.H.; Almars, A.I.; Alelwani, W.; Toraih, E.A.; Nemr, N.A.; Shaalan, A.A.M.; Fawzy, M.S.; Attallah, S.M. Genetic Variation in DEAD-Box Helicase 20 as a Putative Marker of Recurrence in Propensity-Matched Colon Cancer Patients. Genes 2022, 13, 1404. [Google Scholar] [CrossRef]
- Al Ageeli, E.; Attallah, S.M.; Mohamed, M.H.; Almars, A.I.; Kattan, S.W.; Toraih, E.A.; Fawzy, M.S.; Darwish, M.K. Migration/Differentiation-Associated LncRNA. Genes 2022, 13, 1996. [Google Scholar] [CrossRef]
- Li, L.; Sun, R.; Liang, Y.; Pan, X.; Li, Z.; Bai, P.; Zeng, X.; Zhang, D.; Zhang, L.; Gao, L. Association between polymorphisms in long non-coding RNA PRNCR1 in 8q24 and risk of colorectal cancer. J. Exp. Clin. Cancer Res. 2013, 32, 104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiu, Z.; Tian, G.; Zhu, Q.; Zhang, Z.; Qin, R.; Peng, Y.; Tang, W.; Zhang, S.; Xi, Y. Association between long non-coding RNA rs944289 and rs7990916 polymorphisms and the risk of colorectal cancer in a Chinese population. Sci. Rep. 2022, 12, 2495. [Google Scholar] [CrossRef]
- Abdi, E.; Latifi-Navid, S. Long non-coding RNA polymorphisms and hepatocellular carcinoma and pancreatic cancer risk. Pers. Med. 2023, 20, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Han, H.; Choi, S.E.; Park, H.; Woo, H.Y.; Jang, M.; Shim, H.S.; Hwang, S.; Kang, H.; Cho, N.H. p53 Immunohistochemistry and Mutation Types Mismatching in High-Grade Serous Ovarian Cancer. Diagnostics 2022, 12, 579. [Google Scholar] [CrossRef]
- Liu, S.; Wu, M.; Wang, F. Research Progress in Prognostic Factors and Biomarkers of Ovarian Cancer. J. Cancer 2021, 12, 3976–3996. [Google Scholar] [CrossRef]
- Mathieson, W.; Thomas, G. Using FFPE Tissue in Genomic Analyses: Advantages, Disadvantages and the Role of Biospecimen Science. Curr. Pathobiol. Rep. 2019, 7, 35–40. [Google Scholar] [CrossRef]
- Ho, X.D.; Phung, P.; Le, V.Q.; Nguyen, V.H.; Reimann, E.; Prans, E.; Koks, G.; Maasalu, K.; Le, N.T.; Trinh, L.H.; et al. Whole transcriptome analysis identifies differentially regulated networks between osteosarcoma and normal bone samples. Exp. Biol. Med. 2017, 242, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Levin, Y.; Talsania, K.; Tran, B.; Shetty, J.; Zhao, Y.; Mehta, M. Optimization for Sequencing and Analysis of Degraded FFPE-RNA Samples. J. Vis. Exp. JoVE 2020, 160, e61060. [Google Scholar] [CrossRef]
- Liu, Y.; Bhagwate, A.; Winham, S.J.; Stephens, M.T.; Harker, B.W.; McDonough, S.J.; Stallings-Mann, M.L.; Heinzen, E.P.; Vierkant, R.A.; Hoskin, T.L.; et al. Quality control recommendations for RNASeq using FFPE samples based on pre-sequencing lab metrics and post-sequencing bioinformatics metrics. BMC Med. Genom. 2022, 15, 195. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Leskela, S.; Mies, B.P.; Velasco, A.P.; Palacios, J. Morphological and molecular heterogeneity of epithelial ovarian cancer: Therapeutic implications. Eur. J. Cancer Suppl. 2020, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Xu, X.; Ye, M.; Sheng, B.; Zhu, X. The prognostic value of HER2 in ovarian cancer: A meta-analysis of observational studies. PLoS ONE 2018, 13, e0191972. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Jancik, S.; Drabek, J.; Radzioch, D.; Hajduch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef]
- Therachiyil, L.; Anand, A.; Azmi, A.; Bhat, A.; Korashy, H.M.; Uddin, S. Role of RAS signaling in ovarian cancer. F1000Research 2022, 11, 1253. [Google Scholar] [CrossRef]
- Mayr, D.; Hirschmann, A.; Lohrs, U.; Diebold, J. KRAS and BRAF mutations in ovarian tumors: A comprehensive study of invasive carcinomas, borderline tumors and extraovarian implants. Gynecol. Oncol. 2006, 103, 883–887. [Google Scholar] [CrossRef]
- Zhan, L.; Li, J.; Wei, B. Long non-coding RNAs in ovarian cancer. J. Exp. Clin. Cancer Res. 2018, 37, 120. [Google Scholar] [CrossRef]
- Yan, H.; Bu, P. Non-coding RNA in cancer. Essays Biochem. 2021, 65, 625–639. [Google Scholar] [CrossRef]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long non-coding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef]
- Gao, N.; Li, Y.; Li, J.; Gao, Z.; Yang, Z.; Liu, H.; Fan, T. Long Non-Coding RNAs: The Regulatory Mechanisms, Research Strategies, and Future Directions in Cancers. Front. Oncol. 2020, 10, 598817. [Google Scholar] [CrossRef] [PubMed]
- Braga, E.A.; Fridman, M.V.; Moscovtsev, A.A.; Filippova, E.A.; Dmitriev, A.A.; Kushlinskii, N.E. LncRNAs in Ovarian Cancer Progression, Metastasis, and Main Pathways: ceRNA and Alternative Mechanisms. Int. J. Mol. Sci. 2020, 21, 8855. [Google Scholar] [CrossRef] [PubMed]
- Permuth-Wey, J.; Lawrenson, K.; Shen, H.C.; Velkova, A.; Tyrer, J.P.; Chen, Z.; Lin, H.Y.; Chen, Y.A.; Tsai, Y.Y.; Qu, X.; et al. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat. Commun. 2013, 4, 1627. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Shang, X.; Shi, Y.; Yang, Z.; Zhao, J.; Yang, M.; Li, Y.; Xu, S. Genetic variants of lncRNA HOTAIR and risk of epithelial ovarian cancer among Chinese women. Oncotarget 2016, 7, 41047–41052. [Google Scholar] [CrossRef] [PubMed]
- Aznaourova, M.; Schmerer, N.; Schmeck, B.; Schulte, L.N. Disease-Causing Mutations and Rearrangements in Long Non-coding RNA Gene Loci. Front. Genet. 2020, 11, 527484. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Sood, A.K.; Dang, C.V.; Zhang, L. The role of long non-coding RNAs in cancer: The dark matter matters. Curr. Opin. Genet. Dev. 2018, 48, 8–15. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Azimi, T.; Taheri, M. Myocardial Infarction Associated Transcript (MIAT): Review of its impact in the tumorigenesis. Biomed. Pharmacother. 2021, 133, 111040. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, X.; Qian, B.; Feng, N.; Gao, S.; Zhao, Y.; Zhou, B. The lncRNA myocardial infarction associated transcript-centric competing endogenous RNA network in non-small-cell lung cancer. Cancer Manag. Res. 2018, 10, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Luo, C.; Guo, Q.; Cao, J.; Yang, Q.; Dong, K.; Wang, S.; Wang, K.; Song, C. Association analyses of genetic variants in long non-coding RNA MALAT1 with breast cancer susceptibility and mRNA expression of MALAT1 in Chinese Han population. Gene 2018, 642, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Xiang, J.J.; Wu, L.G.; Bai, Y.S.; Chen, Z.W.; Yin, X.Q.; Wang, Q.; Guo, W.H.; Peng, Y.; Guo, H.; et al. A genetic variant in long non-coding RNA MALAT1 associated with survival outcome among patients with advanced lung adenocarcinoma: A survival cohort analysis. BMC Cancer 2017, 17, 167. [Google Scholar] [CrossRef] [PubMed]
- Andrii, V.; Yaroslav, C.; Viktoriia, H.; Alexander, A. Analysis of association between Rs3200401 long non-coding RNA MALAT1 gene polymorphism and prostate adenocarcinoma development in Ukrainian population. J. Urol. Nephrol. Stud. 2019, 1, 99–102. [Google Scholar]
- Ding, Y.F.; Wen, Y.C.; Chuang, C.Y.; Lin, C.W.; Yang, Y.C.; Liu, Y.F.; Chang, W.M.; Chang, L.C.; Yang, S.F.; Chien, M.H. Combined Impacts of Genetic Variants of Long Non-Coding RNA MALAT1 and the Environmental Carcinogen on the Susceptibility to and Progression of Oral Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 684941. [Google Scholar] [CrossRef]
- Qu, Y.; Shao, N.; Yang, W.; Wang, J.; Cheng, Y. Association of polymorphisms in MALAT1 with the risk of esophageal squamous cell carcinoma in a Chinese population. Onco Targets Ther. 2019, 12, 2495–2503. [Google Scholar] [CrossRef]
- Hong, J.H.; Jin, E.H.; Chang, I.A.; Kang, H.; Lee, S.I.; Sung, J.K. Association of long noncoding RNA MALAT1 polymorphisms with gastric cancer risk in Korean individuals. Mol. Genet. Genom. Med. 2020, 8, e1541. [Google Scholar] [CrossRef]
- Ji, X.; Zhang, J.; Liu, L.; Lin, Z.; Pi, L.; Tian, N.; Lin, X.; Chen, S.; Yu, X.; Gao, Y. Association of tagSNPs at lncRNA MALAT-1 with HCC Susceptibility in a Southern Chinese Population. Sci. Rep. 2019, 9, 10895. [Google Scholar] [CrossRef]
- Yuan, L.T.; Chang, J.H.; Lee, H.L.; Yang, Y.C.; Su, S.C.; Lin, C.L.; Yang, S.F.; Chien, M.H. Genetic Variants of lncRNA. J. Clin. Med. 2019, 8, 1406. [Google Scholar] [CrossRef]
- Sun, Y.H.; Chou, Y.H.; Tsai, H.Y.; Hsiao, Y.H.; Lee, C.Y.; Yang, S.F.; Ting, K.H.; Wang, P.H. Impact of Genetic Variants of Long Non-coding RNA. J. Cancer 2022, 13, 2150–2158. [Google Scholar] [CrossRef]
- Li, K.; Han, Z.; Wu, J.; Ye, H.; Sun, G.; Shi, J.; Zhang, J.; Wang, P. The Relationship between. Medicina 2022, 58, 176. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Yan, G.; Yu, S.; Li, F.; Su, Z.; Hou, X.; Xiao, J.; Tian, T. Associations of MALAT1 and its functional single nucleotide polymorphisms with cancer. Pathol. Res. Pract. 2022, 236, 153988. [Google Scholar] [CrossRef] [PubMed]
- Reimann, E.; Koks, S.; Ho, X.D.; Maasalu, K.; Martson, A. Whole exome sequencing of a single osteosarcoma case--integrative analysis with whole transcriptome RNA-seq data. Hum. Genom. 2014, 8, 20. [Google Scholar] [CrossRef]
- Ho, X.D.; Nguyen, H.G.; Trinh, L.H.; Reimann, E.; Prans, E.; Koks, G.; Maasalu, K.; Le, V.Q.; Nguyen, V.H.; Le, N.T.N.; et al. Analysis of the Expression of Repetitive DNA Elements in Osteosarcoma. Front. Genet. 2017, 8, 193. [Google Scholar] [CrossRef]
- Felicio, P.S.; Grasel, R.S.; Campacci, N.; de Paula, A.E.; Galvao, H.C.R.; Torrezan, G.T.; Sabato, C.S.; Fernandes, G.C.; Souza, C.P.; Michelli, R.D.; et al. Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum. Mutat. 2021, 42, 290–299. [Google Scholar] [CrossRef] [PubMed]
- De Marco, C.; Zoppoli, P.; Rinaldo, N.; Morganella, S.; Morello, M.; Zuccala, V.; Carriero, M.V.; Malanga, D.; Chirillo, R.; Bruni, P.; et al. Genome-wide analysis of copy number alterations led to the characterisation of PDCD10 as oncogene in ovarian cancer. Transl. Oncol. 2021, 14, 101013. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.K.; Brambs, C.E.; Hiller, G.G.R.; May, D.; Schmoeckel, E.; Horn, L.C. 2020 WHO Classification of Female Genital Tumors. Geburtshilfe Frauenheilkd 2021, 81, 1145–1153. [Google Scholar] [CrossRef]
- Berek, J.S.; Renz, M.; Kehoe, S.; Kumar, L.; Friedlander, M. Cancer of the ovary, fallopian tube, and peritoneum: 2021 update. Int. J. Gynaecol. Obstet. 2021, 155 (Suppl. S1), 61–85. [Google Scholar] [CrossRef]
- Quan, C.; Ping, J.; Lu, H.; Zhou, G.; Lu, Y. 3DSNP 2.0: Update and expansion of the non-coding genomic variant annotation database. Nucleic Acids Res. 2022, 50, D950–D955. [Google Scholar] [CrossRef]
- Lu, H.; Ma, L.; Quan, C.; Li, L.; Lu, Y.; Zhou, G.; Zhang, C. RegVar: Tissue-specific Prioritization of Noncoding Regulatory Variants. Genom. Proteom. Bioinform. 2021, 21, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Mohammad, H.M.F.; Abdelghany, A.A.; Al Ageeli, E.; Kattan, S.W.; Hassan, R.; Toraih, E.A.; Fawzy, M.S.; Mokhtar, N. Long Non-Coding RNAs Gene Variants as Molecular Markers for Diabetic Retinopathy Risk and Response to Anti-VEGF Therapy. Pharmgenom. Pers. Med. 2021, 14, 997–1014. [Google Scholar] [CrossRef] [PubMed]
- Shaalan, A.A.M.; Mokhtar, S.H.; Ahmedah, H.T.; Almars, A.I.; Toraih, E.A.; Ibrahiem, A.T.; Fawzy, M.S.; Salem, M.A. Prognostic Value of. Biomolecules 2022, 12, 569. [Google Scholar] [CrossRef]
- Elwazir, M.Y.; Hussein, M.H.; Toraih, E.A.; Al Ageeli, E.; Esmaeel, S.E.; Fawzy, M.S.; Faisal, S. Association of Angio-LncRNAs MIAT rs1061540/MALAT1 rs3200401 Molecular Variants with Gensini Score in Coronary Artery Disease Patients Undergoing Angiography. Biomolecules 2022, 12, 137. [Google Scholar] [CrossRef]
- Fawzy, M.S.; Hussein, M.H.; Abdelaziz, E.Z.; Yamany, H.A.; Ismail, H.M.; Toraih, E.A. Association of MicroRNA-196a2 Variant with Response to Short-Acting β2-Agonist in COPD: An Egyptian Pilot Study. PLoS ONE 2016, 11, e0152834. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Total Tumors (N = 182) | Low-Grade Epithelial Tumors (N = 85) | Malignant Epithelial Tumors (N = 97) | p-Value |
|---|---|---|---|---|
| Demographic data | ||||
| Age in years | ||||
| <30 years | 34 (18.7) | 26 (30.6) | 8 (8.2) | 0.001 |
| 30–49 years | 96 (52.7) | 38 (44.7) | 58 (59.8) | |
| ≥50 years | 52 (28.6) | 31 (32.0) | 21 (24.7) | |
| Pathological subtype | ||||
| Benign epithelial tumors | 72 (39.6) | 72 (84.7) | 0 (0.0) | <0.001 |
| Borderline epithelial tumors | 13 (7.1) | 13 (15.3) | 0 (0.0) | |
| High-grade serous carcinoma | 49 (26.9) | 0 (0.0) | 49 (50.5) | |
| Other than high-grade serous | 48 (26.4) | 0 (0.0) | 48 (49.5) | |
| Immunohistochemistry staining | ||||
| HER2 protein staining | ||||
| Negative | 97 (53.3) | 74 (87.1) | 23 (23.7) | <0.001 |
| 1+ | 24 (13.2) | 6 (7.1) | 18 (18.6) | |
| 2+ | 27 (14.8) | 4 (4.7) | 23 (23.7) | |
| 3+ | 34 (18.7) | 1 (1.2) | 33 (34) | |
| P53 protein staining | ||||
| Negative | 134 (73.6) | 85 (100) | 49 (50.5) | <0.001 |
| Positive * | 48 (26.4) | 0 (0.0) | 48 (49.5) | |
| KRAS protein staining | ||||
| Negative | 97 (53.3) | 35 (41.2) | 62 (63.9) | 0.003 |
| Positive | 85 (46.7) | 50 (58.8) | 35 (36.1) | |
| EGFR protein staining | ||||
| Negative | 175 (96.2) | 83 (97.6) | 92 (94.8) | 0.45 |
| Positive | 7 (3.8) | 2 (2.4) | 5 (5.2) | |
| Gene mutation screening | ||||
| BRAF V600 | ||||
| Wild | 16 (8.8) | 9 (10.6) | 7 (7.2) | 0.24 |
| Heterozygote | 154 (84.6) | 73 (85.9) | 81 (83.5) | |
| Mutant | 12 (6.6) | 3 (3.5) | 9 (9.3) | |
| KRAS exon 12 | ||||
| Wild | 28 (15.4) | 15 (17.6) | 13 (13.4) | 0.54 |
| Mutant | 154 (84.6) | 70 (82.4) | 84 (86.6) | |
| KRAS exon 13 | ||||
| Wild | 182 (100) | 85 (100) | 97 (100) | NA |
| Characteristics | Levels | High-Grade Serous Carcinoma (N = 49) | Other than High-Grade Serous (N = 48) | p-Value |
|---|---|---|---|---|
| Demographic data | ||||
| Age in years | <30 years | 2 (4.1) | 6 (12.5) | 0.27 |
| 30–49 years | 32 (65.3) | 2 (54.2) | ||
| ≥50 years | 15 (30.6) | 16 (33.3) | ||
| Immunohistochemistry staining | ||||
| HER2 protein staining | Negative | 11 (22.4) | 12 (25) | 0.28 |
| 1+ | 8 (16.3) | 10 (20.8) | ||
| 2+ | 9 (18.4) | 14 (29.2) | ||
| 3+ | 21 (42.9) | 12 (25) | ||
| P53 protein staining | Negative | 0 (0.0) | 48 (100) | <0.001 |
| Positive * | 49 (100) | 0 (0.0) | ||
| KRAS protein staining | Negative | 30 (61.2) | 32 (66.7) | 0.67 |
| Positive | 19 (38.8) | 16 (33.3) | ||
| EGFR protein staining | Negative | 45 (91.8) | 47 (97.9) | 0.36 |
| Positive | 4 (8.2) | 1 (2.1) | ||
| Gene mutation | ||||
| BRAF V600 | Wild | 1 (2.1) | 6 (12.2) | 0.15 |
| Heterozygote | 42 (87.5) | 39 (79.6) | ||
| Mutant | 5 (10.4) | 4 (8.2) | ||
| KRAS exon 12 | Wild | 11 (22.9) | 2 (4.1) | 0.007 |
| Mutant | 37 (77.1) | 47 (95.9) | ||
| KRAS exon 13 | Wild | 49 (100) | 48 (100) | NA |
| Model | Genotype | Low-Grade Epithelial Tumors (N = 85) | Malignant Epithelial Tumors (N = 97) | OR (95% CI) | p-Value | AIC | BIC |
|---|---|---|---|---|---|---|---|
| Codominant | C/C | 31 (36.5%) | 35 (36.1%) | 1 | 0.43 | 251 | 263.9 |
| C/T | 43 (50.6%) | 44 (45.4%) | 0.93 (0.48–1.79) | ||||
| T/T | 11 (12.9%) | 18 (18.6%) | 1.64 (0.66–4.09) | ||||
| Dominant | C/C | 31 (36.5%) | 35 (36.1%) | 1 | 0.83 | 250.7 | 260.3 |
| C/T-T/T | 54 (63.5%) | 62 (63.9%) | 1.07 (0.58–1.98) | ||||
| Recessive | C/C-C/T | 74 (87.1%) | 79 (81.4%) | 1 | 0.20 | 249.1 | 258.7 |
| T/T | 11 (12.9%) | 18 (18.6%) | 1.71 (0.74–3.93) | ||||
| Overdominant | C/C-T/T | 42 (49.4%) | 53 (54.6%) | 1 | 0.46 | 250.2 | 259.8 |
| C/T | 43 (50.6%) | 44 (45.4%) | 0.80 (0.44–1.45) |
| Model | Genotype | Low-Grade Epithelial Tumors | Malignant Epithelial Tumors | OR (95% CI) | p-Value | AIC | BIC |
|---|---|---|---|---|---|---|---|
| Codominant | C/C | 32 (37.6%) | 55 (56.7%) | 1 | 0.040 | 246.3 | 259.1 |
| C/T | 31 (36.5%) | 27 (27.8%) | 0.53 (0.27–1.05) | ||||
| T/T | 22 (25.9%) | 15 (15.5%) | 0.39 (0.18–0.88) | ||||
| Dominant | C/C | 32 (37.6%) | 55 (56.7%) | 1 | 0.014 | 244.7 | 254.3 |
| C/T-T/T | 53 (62.4%) | 42 (43.3%) | 0.47 (0.26–0.87) | ||||
| Recessive | C/C-C/T | 63 (74.1%) | 82 (84.5%) | 1 | 0.08 | 247.6 | 257.2 |
| T/T | 22 (25.9%) | 15 (15.5%) | 0.51 (0.24–1.08) | ||||
| Overdominant | C/C-T/T | 54 (63.5%) | 70 (72.2%) | 1 | 0.28 | 249.5 | 259.2 |
| C/T | 31 (36.5%) | 27 (27.8%) | 0.70 (0.37–1.33) |
| MALAT1 | MIAT | Total | Low-Grade Epithelial Tumors | Malignant Epithelial Tumors | OR (95% CI) | p-Value | |
|---|---|---|---|---|---|---|---|
| 1 | C | C | 0.407 | 0.4 | 0.423 | 1 | --- |
| 2 | T | C | 0.23 | 0.159 | 0.283 | 1.67 (0.90–3.11) | 0.11 |
| 3 | C | T | 0.195 | 0.217 | 0.164 | 0.78 (0.45–1.37) | 0.39 |
| 4 | T | T | 0.168 | 0.224 | 0.13 | 0.65 (0.36–1.20) | 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fawzy, M.S.; Ibrahiem, A.T.; Osman, D.M.; Almars, A.I.; Alshammari, M.S.; Almazyad, L.T.; Almatrafi, N.D.A.; Almazyad, R.T.; Toraih, E.A. Angio-Long Noncoding RNA MALAT1 (rs3200401) and MIAT (rs1061540) Gene Variants in Ovarian Cancer. Epigenomes 2024, 8, 5. https://doi.org/10.3390/epigenomes8010005
Fawzy MS, Ibrahiem AT, Osman DM, Almars AI, Alshammari MS, Almazyad LT, Almatrafi NDA, Almazyad RT, Toraih EA. Angio-Long Noncoding RNA MALAT1 (rs3200401) and MIAT (rs1061540) Gene Variants in Ovarian Cancer. Epigenomes. 2024; 8(1):5. https://doi.org/10.3390/epigenomes8010005
Chicago/Turabian StyleFawzy, Manal S., Afaf T. Ibrahiem, Dalia Mohammad Osman, Amany I. Almars, Maali Subhi Alshammari, Layan Tariq Almazyad, Noof Daif Allah Almatrafi, Renad Tariq Almazyad, and Eman A. Toraih. 2024. "Angio-Long Noncoding RNA MALAT1 (rs3200401) and MIAT (rs1061540) Gene Variants in Ovarian Cancer" Epigenomes 8, no. 1: 5. https://doi.org/10.3390/epigenomes8010005
APA StyleFawzy, M. S., Ibrahiem, A. T., Osman, D. M., Almars, A. I., Alshammari, M. S., Almazyad, L. T., Almatrafi, N. D. A., Almazyad, R. T., & Toraih, E. A. (2024). Angio-Long Noncoding RNA MALAT1 (rs3200401) and MIAT (rs1061540) Gene Variants in Ovarian Cancer. Epigenomes, 8(1), 5. https://doi.org/10.3390/epigenomes8010005