
Epigenetic Mechanisms in Hematologic Aging and Premalignant Conditions

Abstract
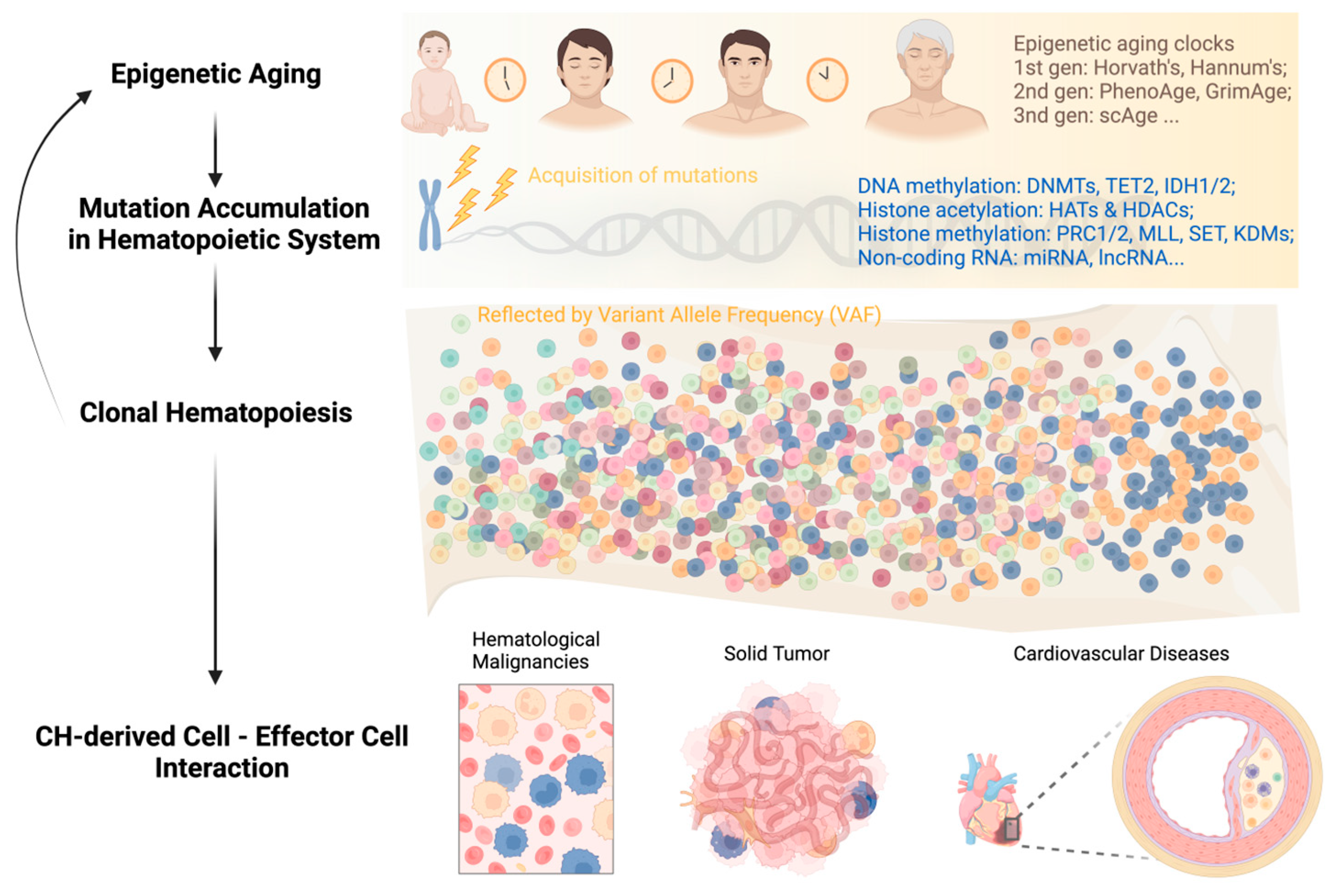
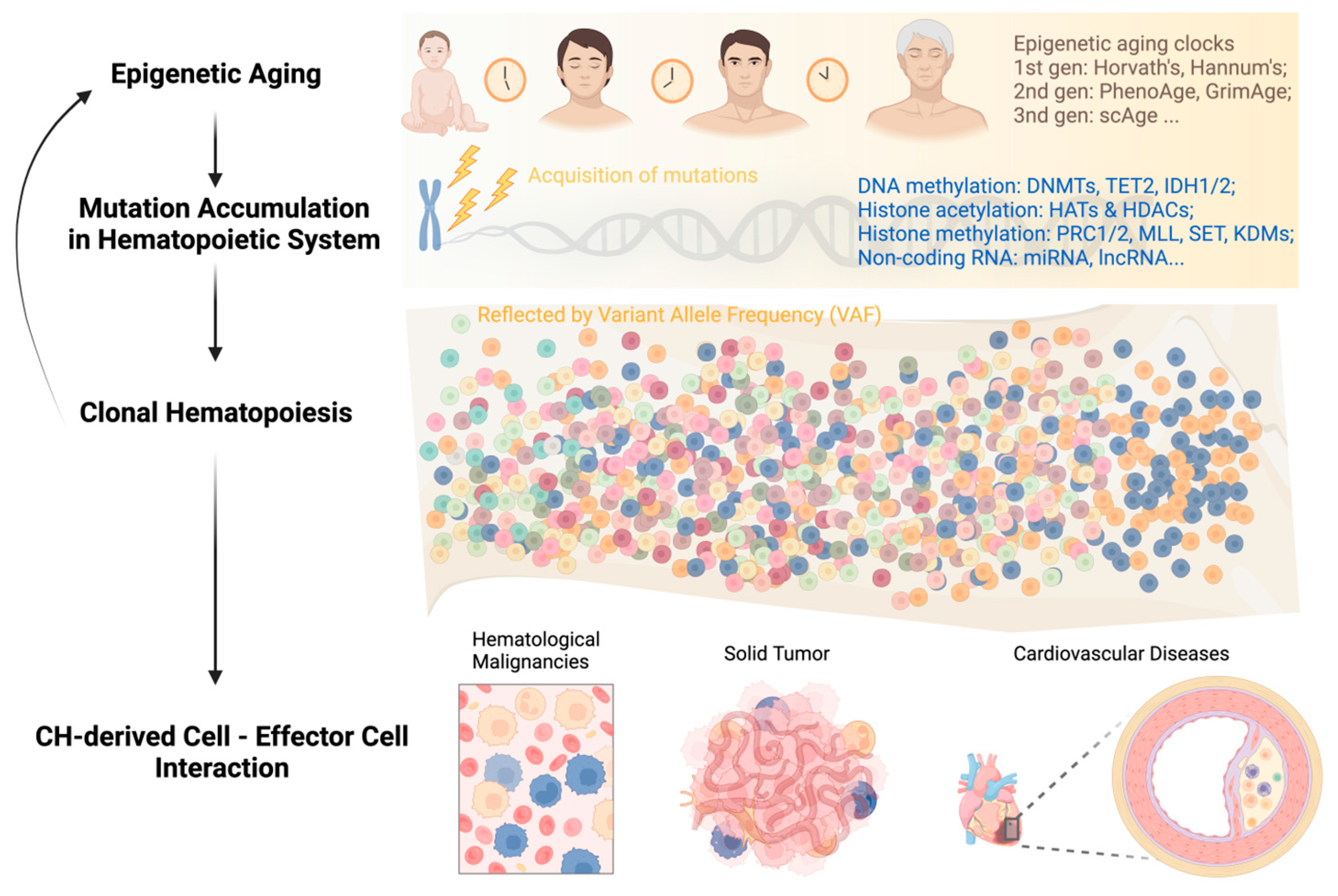
:1. Introduction
2. Epigenetic Regulation of HSCs
2.1. DNA Methylation
2.2. Histone Acetylation
2.3. Histone Methylation
2.4. Noncoding RNAs
2.5. Interplay of Epigenetic Regulators
3. Alterations to the Epigenome in HSCs Aging
3.1. Imbalance of Histone Modifications
3.2. Age-Related DNA Methylation Changes and Epigenetic Clocks
4. Epigenetics in Acquisition of Clonal Hematopoiesis with Age
5. Strategies to Alleviate Aging
5.1. Caloric or Dietary Restriction
5.2. Small Molecule-Based Therapy
5.3. Gene Expression Regulation
5.4. Epigenetic Reprogramming
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Buisman, S.C.; de Haan, G. Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies. Cells 2019, 8, 868. [Google Scholar] [CrossRef] [PubMed]
- Akunuru, S.; Geiger, H. Aging, Clonality, and Rejuvenation of Hematopoietic Stem Cells. Trends Mol. Med. 2016, 22, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA Methyltransferase 1 Is Essential for and Uniquely Regulates Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.Q.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.C.; Xi, Y.X.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, U23–U43. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.Q.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.B.; Xia, Z.; et al. Dnmt3a and Dnmt3b Have Overlapping and Distinct Functions in Hematopoietic Stem Cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.J.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef]
- Li, Z.; Cai, X.Q.; Cai, C.L.; Wang, J.P.; Zhang, W.Y.; Petersen, B.E.; Yang, F.C.; Xu, M.J. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Quivoron, C.; Couronne, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 276. [Google Scholar] [CrossRef]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Duy, C.; Beguelin, W.; Melnick, A. Epigenetic Mechanisms in Leukemias and Lymphomas. Cold Spring Harb. Perspect. Med. 2020, 10, a034959. [Google Scholar] [CrossRef]
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Cote, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef] [PubMed]
- Khokhar, E.S.; Borikar, S.; Eudy, E.; Stearns, T.; Young, K.; Trowbridge, J.J. Aging-associated decrease in the histone acetyltransferase KAT6B is linked to altered hematopoietic stem cell differentiation. Exp. Hematol. 2020, 82, 43–52. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Yang, Y.Q.; Schreuder, J.; Nilsson, S.K.; Bilardi, R.; Carotta, S.; McRae, H.M.; Metcalf, D.; Voss, A.K.; Thomas, T. MOZ (KAT6A) is essential for the maintenance of classically defined adult hematopoietic stem cells. Blood 2016, 128, 2307–2318. [Google Scholar] [CrossRef]
- Katsumoto, T.; Aikawa, Y.; Iwama, A.; Ueda, S.; Ichikawa, H.; Ochiya, T.; Kitabayashi, I. MOZ is essential for maintenance of hematopoietic stem cells. Gene Dev. 2006, 20, 1321–1330. [Google Scholar] [CrossRef]
- Valerio, D.G.; Xu, H.M.; Eisold, M.E.; Woolthuis, C.M.; Pandita, T.K.; Armstrong, S.A. Histone acetyltransferase activity of MOF is required for adult but not early fetal hematopoiesis in mice. Blood 2017, 129, 48–59. [Google Scholar] [CrossRef]
- Zhang, L.; Mack, R.; Breslin, P.; Zhang, J.W. Molecular and cellular mechanisms of aging in hematopoietic stem cells and their niches. J. Hematol. Oncol. 2020, 13, 157. [Google Scholar] [CrossRef] [PubMed]
- Perez-Campo, F.M.; Borrow, J.; Kouskoff, V.; Lacaud, G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood 2009, 113, 4866–4874. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.I.; Hannah, R.L.; Dawson, M.A.; Pridans, C.; Foster, D.; Joshi, A.; Gottgens, B.; Van Deursen, J.M.; Huntly, B.J.P. The Transcriptional Coactivator Cbp Regulates Self-Renewal and Differentiation in Adult Hematopoietic Stem Cells. Mol. Cell Biol. 2011, 31, 5046–5060. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.L.; Sutton, S.E.; Pletcher, M.T.; Wiltshire, T.; Tarantino, L.M.; Hogenesch, J.B.; Cooke, M.P. c-Myb and p300 regulate stem cell proliferation an hematopoietic differentiation. Dev. Cell 2005, 8, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Rebel, V.I.; Kung, A.L.; Tanner, E.A.; Yang, H.; Bronson, R.T.; Livingston, D.M. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2002, 99, 14789–14794. [Google Scholar] [CrossRef] [PubMed]
- Man, N.; Mas, G.; Karl, D.L.; Sun, J.; Liu, F.; Yang, Q.; Torres-Martin, M.; Itonaga, H.; Martinez, C.; Chen, S.; et al. p300 suppresses the transition of myelodysplastic syndromes to acute myeloid leukemia. JCI Insight 2021, 6, e138478. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Heideman, M.R.; Lancini, C.; Proost, N.; Yanover, E.; Jacobs, H.; Dannenberg, J.H. Sin3a-associated Hdac1 and Hdac2 are essential for hematopoietic stem cell homeostasis and contribute differentially to hematopoiesis. Haematologica 2014, 99, 1292–1303. [Google Scholar] [CrossRef]
- Wada, T.; Kikuchi, J.; Nishimura, N.; Shimizu, R.; Kitamura, T.; Furukawa, Y. Expression levels of histone deacetylases determine the cell fate of hematopoietic progenitors. J. Biol. Chem. 2009, 284, 30673–30683. [Google Scholar] [CrossRef]
- Elizalde, C.; Fernandez-Rueda, J.; Salcedo, J.M.; Dorronsoro, A.; Ferrin, I.; Jakobsson, E.; Trigueros, C. Histone deacetylase 3 modulates the expansion of human hematopoietic stem cells. Stem Cells Dev. 2012, 21, 2581–2591. [Google Scholar] [CrossRef]
- Summers, A.R.; Fischer, M.A.; Stengel, K.R.; Zhao, Y.; Kaiser, J.F.; Wells, C.E.; Hunt, A.; Bhaskara, S.; Luzwick, J.W.; Sampathi, S.; et al. HDAC3 is essential for DNA replication in hematopoietic progenitor cells. J. Clin. Investig. 2013, 123, 3112–3123. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.K.; Qi, J.; Cai, Q.; Carnahan, E.; Ayala Ramirez, M.; Li, L.; Marcucci, G.; Kuo, Y.H. HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood 2017, 130, 2619–2630. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Diao, D.J.; Shi, Z.C.; Zhu, X.D.; Gao, Y.W.; Gao, S.R.; Liu, X.Y.; Wu, Y.; Rudolph, K.L.; Liu, G.H.; et al. SIRT6 Controls Hematopoietic Stem Cell Homeostasis through Epigenetic Regulation of Wnt Signaling. Cell Stem Cell 2016, 18, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Iwama, A.; Oguro, H.; Negishi, M.; Kato, Y.; Morita, Y.; Tsukui, H.; Ema, H.; Kamijo, T.; Katoh-Fukui, Y.; Koseki, H.; et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 2004, 21, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I.; Herrera-Merchan, A.; Ligos, J.M.; Carramolino, L.; Nunez, J.; Martinez, F.; Dominguez, O.; Torres, M.; Gonzalez, S. Ezh1 Is Required for Hematopoietic Stem Cell Maintenance and Prevents Senescence-like Cell Cycle Arrest. Cell Stem Cell 2012, 11, 649–662. [Google Scholar] [CrossRef]
- Kamminga, L.M.; Bystrykh, L.V.; Boer, A.C.; Houwer, S.; Douma, J.; Weersing, E.; Dontje, B.; de Haan, G. The polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 2006, 107, 2170–2179. [Google Scholar] [CrossRef]
- Lee, S.C.W.; Miller, S.; Hyland, C.; Kauppi, M.; Lebois, M.; Di Rago, L.; Metcalf, D.; Kinkel, S.A.; Josefsson, E.C.; Blewitt, M.E.; et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 2015, 126, 167–175. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Ding, Y.; Yao, Y.; Gong, X.; Zhuo, Q.; Chen, J.; Tian, M.; Farzaneh, M. JMJD3: A critical epigenetic regulator in stem cell fate. Cell Commun. Signal 2021, 19, 72. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Thieme, S.; Gyarfas, T.; Richter, C.; Ozhan, G.; Fu, J.; Alexopoulou, D.; Muders, M.H.; Michalk, I.; Jakob, C.; Dahl, A.; et al. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 2013, 121, 2462–2473. [Google Scholar] [CrossRef] [PubMed]
- Sera, Y.; Nakata, Y.; Ueda, T.; Yamasaki, N.; Koide, S.; Kobayashi, H.; Ikeda, K.; Kobatake, K.; Iwasaki, M.; Oda, H.; et al. UTX maintains the functional integrity of the murine hematopoietic system by globally regulating aging-associated genes. Blood 2021, 137, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef]
- Mallaney, C.; Ostrander, E.L.; Celik, H.; Kramer, A.C.; Martens, A.; Kothari, A.; Koh, W.K.; Haussler, E.; Iwamori, N.; Gontarz, P.; et al. Kdm6b regulates context-dependent hematopoietic stem cell self-renewal and leukemogenesis. Leukemia 2019, 33, 2506–2521. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ernst, P. Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis. Curr. Opin. Hematol. 2017, 24, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Fisher, J.K.; Avery, W.; Wade, S.; Foy, D.; Korsmeyer, S.J. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev. Cell 2004, 6, 437–443. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 Is a Haploinsufficient 7q Tumor Suppressor in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef]
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111. [Google Scholar] [CrossRef]
- Tusi, B.K.; Deng, C.W.; Salz, T.; Zeumer, L.; Li, Y.Q.; So, C.W.E.; Morel, L.M.; Qiu, Y.; Huang, S.M. Setd1a regulates progenitor B-cell-to-precursor B-cell development through histone H3 lysine 4 trimethylation and Ig heavy-chain rearrangement. Faseb J. 2015, 29, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.H.; Albert, M.; Sroczynska, P.; Cruickshank, V.A.; Guo, Y.P.; Rossi, D.J.; Helin, K.; Enver, T. The histone demethylase Jarid1b is required for hematopoietic stem cell self-renewal in mice. Blood 2015, 125, 2075–2078. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, M.A.; Shao, Z.; Hsu, Y.J.; Guo, G.; Luc, S.; O’Brien, K.; Fujiwara, Y.; Peng, C.; Nguyen, M.; Orkin, S.H. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife 2013, 2, e00633. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, F.; Sousae, R.; Cinquin, B.; Martin, E.W.; Krietsch, J.; Sanchez, G.; Inman, M.; Tsang, H.; Warr, M.; Passegue, E.; et al. Progressive Chromatin Condensation and H3K9 Methylation Regulate the Differentiation of Embryonic and Hematopoietic Stem Cells. Stem Cell Rep. 2015, 5, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Djeghloul, D.; Kuranda, K.; Kuzniak, I.; Barbieri, D.; Naguibneva, I.; Choisy, C.; Bories, J.C.; Dosquet, C.; Pla, M.; Vanneaux, V.; et al. Age-Associated Decrease of the Histone Methyltransferase SUV39H1 in HSC Perturbs Heterochromatin and B Lymphoid Differentiation. Stem Cell Rep. 2016, 6, 970–984. [Google Scholar] [CrossRef]
- Pasquarella, A.; Nuber, A.; Schotta, G. Deletion of the Histone Methyltransferase Setdb1 during Hematopoiesis Results in Hematopoietic Stem Cell Failure and Abrogates B Cell Development. Exp. Hematol. 2013, 41, S19. [Google Scholar] [CrossRef]
- Koide, S.; Oshima, M.; Takubo, K.; Yamazaki, S.; Nitta, E.; Saraya, A.; Aoyama, K.; Kato, Y.; Miyagi, S.; Nakajima-Takagi, Y.; et al. Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood 2016, 128, 638–649. [Google Scholar] [CrossRef]
- Ortiz, G.G.R.; Mohammadi, Y.; Nazari, A.; Ataeinaeini, M.; Kazemi, P.; Yasamineh, S.; Al-Naqeeb, B.Z.T.; Zaidan, H.K.; Gholizadeh, O. A state-of-the-art review on the MicroRNAs roles in hematopoietic stem cell aging and longevity. Cell Commun. Signal 2023, 21, 85. [Google Scholar] [CrossRef]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. MiR-125 in normal and malignant hematopoiesis. Leukemia 2012, 26, 2011–2018. [Google Scholar] [CrossRef]
- Chung, S.S.; Hu, W.; Park, C.Y. The Role of MicroRNAs in Hematopoietic Stem Cell and Leukemic Stem Cell Function. Ther. Adv. Hematol. 2011, 2, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, E.E.; Broekhuis, M.J.C.; Weersing, E.; Dinitzen, A.; Verovskaya, E.; Ausema, A.; Ritsema, M.; Zwart, E.; de Haan, G.; Bystrykh, L.V. MiR-125a enhances self-renewal, lifespan, and migration of murine hematopoietic stem and progenitor cell clones. Sci. Rep. 2019, 9, 4785. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Jeong, M.; Sun, D.Q.; Park, H.J.; Rodriguez, B.A.T.; Xia, Z.; Yang, L.B.; Zhang, X.T.; Sheng, K.W.; Darlington, G.J.; et al. Long Non-Coding RNAs Control Hematopoietic Stem Cell Function. Cell Stem Cell 2015, 16, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Magilnick, N.; Boldin, M.P. Molecular Moirai: Long Noncoding RNA Mediators of HSC Fate. Curr. Stem Cell Rep. 2018, 4, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Delas, M.J.; Jackson, B.T.; Kovacevic, T.; Vangelisti, S.; Maravilla, E.M.; Wild, S.A.; Stork, E.M.; Erard, N.; Knott, S.R.V.; Hannon, G.J. lncRNA Spehd Regulates Hematopoietic Stem and Progenitor Cells and Is Required for Multilineage Differentiation. Cell Rep. 2019, 27, 719–729.e6. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, R.; Qiu, R.; Zheng, Y.; Huang, W.; Hu, H.; Ji, Q.; He, H.; Shang, Y.; Gong, Y.; et al. CRL4B promotes tumorigenesis by coordinating with SUV39H1/HP1/DNMT3A in DNA methylation-based epigenetic silencing. Oncogene 2015, 34, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef]
- Epsztejn-Litman, S.; Feldman, N.; Abu-Remaileh, M.; Shufaro, Y.; Gerson, A.; Ueda, J.; Deplus, R.; Fuks, F.; Shinkai, Y.; Cedar, H.; et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 2008, 15, 1176–1183. [Google Scholar] [CrossRef]
- Chang, Y.Q.; Sun, L.D.; Kokura, K.; Horton, J.R.; Fukuda, M.; Espejo, A.; Izumi, V.; Koomen, J.M.; Bedford, M.T.; Zhang, X.; et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011, 2, 533. [Google Scholar] [CrossRef]
- Ramabadran, R.; Wang, J.H.; Reyes, J.M.; Guzman, A.G.; Gupta, S.; Rosas, C.; Brunetti, L.; Gundry, M.C.; Tovy, A.; Long, H.; et al. DNMT3A-coordinated splicing governs the stem state switch towards differentiation in embryonic and haematopoietic stem cells. Nat. Cell Biol. 2023, 25, 528–539. [Google Scholar] [CrossRef]
- Rush, M.; Appanah, R.; Lee, S.; Lam, L.L.; Goyal, P.; Lorincz, M.C. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics 2009, 4, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.Q.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.R.; Iannarella, N.; Naselli, G.; Bediaga, N.G.; Johanson, T.M.; Harrison, L.C.; Allan, R.S. Extreme disruption of heterochromatin is required for accelerated hematopoietic aging. Blood 2020, 135, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Rimmele, P.; Bigarella, C.L.; Liang, R.; Izac, B.; Dieguez-Gonzalez, R.; Barbet, G.; Donovan, M.; Brugnara, C.; Blander, J.M.; Sinclair, D.A.; et al. Aging-like phenotype and defective lineage specification in SIRT1-deleted hematopoietic stem and progenitor cells. Stem Cell Rep. 2014, 3, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.K.; Zhao, B.; Lombard, D.B.; Fingar, D.C.; Inoki, K. Cross-talk between Sirtuin and Mammalian Target of Rapamycin Complex 1 ( mTORC1) Signaling in the Regulation of S6 Kinase 1 ( S6K1) Phosphorylation. J. Biol. Chem. 2014, 289, 13132–13141. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Wang, Z.; Chen, W.Y. Sirtuins in hematological aging and malignancy. Crit. Rev. Oncog. 2013, 18, 531–547. [Google Scholar] [CrossRef]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef]
- Petkovich, D.A.; Podolskiy, D.I.; Lobanov, A.V.; Lee, S.G.; Miller, R.A.; Gladyshev, V.N. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab. 2017, 25, 954–960.e6. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging-Us 2018, 10, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.F.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging-Us 2019, 11, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Trapp, A.; Kerepesi, C.; Gladyshev, V. Profiling Epigenetic Age in Single Cells. Innov. Aging 2021, 5, 673. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.C.; Hu, Q.C.; Wang, L.N.; Liu, J.Q.; Zheng, Z.K.; Zhang, W.Q.; Ren, J.; Zhu, F.F.; Liu, G.H. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Tar. 2022, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef]
- Thompson, D.J.; Genovese, G.; Halvardson, J.; Ulirsch, J.C.; Wright, D.J.; Terao, C.; Davidsson, O.B.; Day, F.R.; Sulem, P.; Jiang, Y.; et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 2019, 575, 652–657. [Google Scholar] [CrossRef]
- Loh, P.R.; Genovese, G.; Handsaker, R.E.; Finucane, H.K.; Reshef, Y.A.; Palamara, P.F.; Birmann, B.M.; Talkowski, M.E.; Bakhoum, S.F.; McCarroll, S.A.; et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 2018, 559, 350–355. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; You, X.; Droin, N.; Banaszak, L.G.; Churpek, J.; Padron, E.; Geissler, K.; Solary, E.; Patnaik, M.M.; Zhang, J. Role of ASXL1 in hematopoiesis and myeloid diseases. Exp. Hematol. 2022, 115, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Lee-Six, H.; Obro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.; Jahn, N.; Jaiswal, S. Clonal Hematopoiesis and Its Impact on Human Health. Annu. Rev. Med. 2023, 74, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Buscarlet, M.; Mollica, L.; Levine, R.L. Concise Review: Age-Related Clonal Hematopoiesis: Stem Cells Tempting the Devil. Stem Cells 2018, 36, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.; Spencer Chapman, M.; Williams, N.; Dawson, K.J.; Mende, N.; Calderbank, E.F.; Jung, H.; Mitchell, T.; Coorens, T.H.H.; Spencer, D.H.; et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 2022, 606, 343–350. [Google Scholar] [CrossRef]
- Vijg, J.; Schumacher, B.; Abakir, A.; Antonov, M.; Bradley, C.; Cagan, A.; Church, G.; Gladyshev, V.N.; Gorbunova, V.; Maslov, A.Y.; et al. Mitigating age-related somatic mutation burden. Trends Mol. Med. 2023, 29, 530–540. [Google Scholar] [CrossRef]
- Natarajan, P. Genomic Aging, Clonal Hematopoiesis, and Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2023, 43, 3–14. [Google Scholar] [CrossRef]
- Saadatagah, S.; Ballantyne, C.M. Clonal hematopoiesis of indeterminate potential and cardiovascular disease. Transl. Res. 2023, 255, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, K.; Feng, Y.; Shabashvili, D.; Guryanova, O.A. Alterations to DNMT3A in Hematologic Malignancies. Cancer Res. 2021, 81, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Levine, R.L. TET2 in Normal and Malignant Hematopoiesis. Cold Spring Harb. Perspect. Med. 2017, 7, a026518. [Google Scholar] [CrossRef] [PubMed]
- Vaddavalli, P.L.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022, 38, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Pietrocola, F.; Roiz-Valle, D.; Galluzzi, L.; Kroemer, G. Meta-hallmarks of aging and cancer. Cell Metab. 2023, 35, 12–35. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, S.M.; Cohen, H.J.; Muss, H.; Tew, W.P.; Korc-Grodzicki, B. From Assessment to Implementation and Beyond in Cancer and Aging Research. J. Clin. Oncol. 2021, 39, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Sidlow, R.; Lin, A.E.; Gupta, D.; Jones, L.W.; Moslehi, J.; Zeiher, A.; Jaiswal, S.; Schulz, C.; Blankstein, R.; et al. Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 567–577. [Google Scholar] [CrossRef]
- Evans, M.A.; Sano, S.; Walsh, K. Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu. Rev. Pathol. 2020, 15, 419–438. [Google Scholar] [CrossRef]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4. [Google Scholar] [CrossRef]
- Zekavat, S.M.; Viana-Huete, V.; Matesanz, N.; Jorshery, S.D.; Zuriaga, M.A.; Uddin, M.M.; Trinder, M.; Paruchuri, K.; Zorita, V.; Ferrer-Perez, A.; et al. TP53-mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease. Nat. Cardiovasc. Res. 2023, 2, 144–158. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 (V617F) Mice. Circ. Res. 2018, 123, e35–e47. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.D.M.; Nguyen, N.Q.H.; Yu, B.; Brody, J.A.; Pampana, A.; Nakao, T.; Fornage, M.; Bressler, J.; Sotoodehnia, N.; Weinstock, J.S.; et al. Clonal hematopoiesis of indeterminate potential, DNA methylation, and risk for coronary artery disease. Nat. Commun. 2022, 13, 5350. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.C.; Madar, A.; Campbell, C.D.; He, Y.; Sultan, M.; Healey, M.L.; Xu, H.; D’Aco, K.; Fernandez, A.; Wache-Mainier, C.; et al. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022, 7, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Gumuser, E.D.; Schuermans, A.; Cho, S.M.J.; Sporn, Z.A.; Uddin, M.M.; Paruchuri, K.; Nakao, T.; Yu, Z.; Haidermota, S.; Hornsby, W.; et al. Clonal Hematopoiesis of Indeterminate Potential Predicts Adverse Outcomes in Patients With Atherosclerotic Cardiovascular Disease. J. Am. Coll. Cardiol. 2023, 81, 1996–2009. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brune, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef]
- Tian, R.; Wiley, B.; Liu, J.; Zong, X.; Truong, B.; Zhao, S.; Uddin, M.M.; Niroula, A.; Miller, C.A.; Mukherjee, S.; et al. Clonal Hematopoiesis and Risk of Incident Lung Cancer. J. Clin. Oncol. 2023, 41, 1423–1433. [Google Scholar] [CrossRef]
- Severson, E.A.; Riedlinger, G.M.; Connelly, C.F.; Vergilio, J.A.; Goldfinger, M.; Ramkissoon, S.; Frampton, G.M.; Ross, J.S.; Fratella-Calabrese, A.; Gay, L.; et al. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood 2018, 131, 2501–2505. [Google Scholar] [CrossRef]
- Ptashkin, R.N.; Mandelker, D.L.; Coombs, C.C.; Bolton, K.; Yelskaya, Z.; Hyman, D.M.; Solit, D.B.; Baselga, J.; Arcila, M.E.; Ladanyi, M.; et al. Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. JAMA Oncol. 2018, 4, 1589–1593. [Google Scholar] [CrossRef]
- Marshall, C.H.; Gondek, L.P.; Luo, J.; Antonarakis, E.S. Clonal Hematopoiesis of Indeterminate Potential in Patients with Solid Tumor Malignancies. Cancer Res. 2022, 82, 4107–4113. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.D.; Damask, A.; O’Keeffe, S.; Banerjee, N.; Li, D.; Watanabe, K.; Marketta, A.; Van Meter, M.; Semrau, S.; Horowitz, J.; et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 2022, 612, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.P.; Quiros, P.M.; Gu, M.; Jiang, T.; Mitchell, J.; Langdon, R.; Iyer, V.; Barcena, C.; Vijayabaskar, M.S.; Fabre, M.A.; et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet. 2022, 54, 1155–1166. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Bejar, R. Clonal hematopoiesis in cancer. Exp. Hematol. 2020, 83, 105–112. [Google Scholar] [CrossRef]
- Feng, Y.; Yuan, Q.; Newsome, R.C.; Robinson, T.; Bowman, R.L.; Zuniga, A.N.; Hall, K.N.; Bernsten, C.M.; Shabashvili, D.E.; Krajcik, K.I.; et al. Hematopoietic-specific heterozygous loss of Dnmt3a exacerbates colitis-associated colon cancer. J. Exp. Med. 2023, 220, e20230011. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Comen, E.; Wen, H.Y.; Bastian, L.; Blum, B.; Rapaport, F.T.; Keller, M.; Granot, Z.; Socci, N.; Viale, A.; et al. Somatic mutations in leukocytes infiltrating primary breast cancers. NPJ Breast Cancer 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.A.; Hillary, R.F.; McCartney, D.L.; Terradas-Terradas, M.; Higham, J.; Sproul, D.; Deary, I.J.; Kirschner, K.; Marioni, R.E.; Chandra, T. Age-related clonal haemopoiesis is associated with increased epigenetic age. Curr. Biol. 2019, 29, R786–R787. [Google Scholar] [CrossRef]
- Nachun, D.; Lu, A.T.; Bick, A.G.; Natarajan, P.; Weinstock, J.; Szeto, M.D.; Kathiresan, S.; Abecasis, G.; Taylor, K.D.; Guo, X.; et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 2021, 20, e13366. [Google Scholar] [CrossRef]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 2022, 23, 56–73. [Google Scholar] [CrossRef]
- Fontana, L.; Ghezzi, L.; Cross, A.H.; Piccio, L. Effects of dietary restriction on neuroinflammation in neurodegenerative diseases. J. Exp. Med. 2021, 218, e20190086. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, Y.T.; Wu, J.Y.; Zeng, T.; Cui, H.; Tao, Z.D.; Lei, L.; Yu, L.; Liu, A.W.; Wang, H.; et al. Long-term mid-onset dietary restriction rejuvenates hematopoietic stem cells and improves regeneration capacity of total bone marrow from aged mice. Aging Cell 2020, 19, e13241. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.Z.; Tao, S.; Chen, Z.Y.; Koliesnik, I.O.; Calmes, P.G.; Hoerr, V.; Han, B.; Gebert, N.; Zornig, M.; Loffler, B.; et al. Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J. Exp. Med. 2016, 213, 535–553. [Google Scholar] [CrossRef]
- Lazare, S.; Ausema, A.; Reijne, A.C.; van Dijk, G.; van Os, R.; de Haan, G. Lifelong dietary intervention does not affect hematopoietic stem cell function. Exp. Hematol. 2017, 53, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Dellorusso, P.V.; Verovskaya, E.V.; Bakker, S.T.; Flach, J.; Smith, L.K.; Ventura, P.B.; Lansinger, O.M.; Herault, A.; Zhang, S.Y.; et al. Aged hematopoietic stem cells are refractory to bloodborne systemic rejuvenation interventions. J. Exp. Med. 2021, 218, e20210223. [Google Scholar] [CrossRef]
- Florian, M.C.; Dorr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 Activity Regulates Hematopoietic Stem Cell Aging and Rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Leins, H.; Mulaw, M.; Eiwen, K.; Sakk, V.; Liang, Y.; Denkinger, M.; Geiger, H.; Schirmbeck, R. Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood 2018, 132, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, P.; Gajzer, D.C.; Schaniel, C.; D’Souza, S.; Hoffman, R. Epigenetic reprogramming induces the expansion of cord blood stem cells. J. Clin. Investig. 2014, 124, 2378–2395. [Google Scholar] [CrossRef]
- Zimran, E.; Papa, L.; Djedaini, M.; Patel, A.; Iancu-Rubin, C.; Hoffman, R. Expansion and preservation of the functional activity of adult hematopoietic stem cells cultured ex vivo with a histone deacetylase inhibitor. Stem Cells Transl. Med. 2020, 9, 531–542. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal 2009, 2, ra75. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Tang, W.; Deater, M.; Phan, N.; Marcogliese, A.N.; Li, H.; Al-Dhalimy, M.; Major, A.; Olson, S.; Monnat, R.J., Jr.; et al. Metformin improves defective hematopoiesis and delays tumor formation in Fanconi anemia mice. Blood 2016, 128, 2774–2784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhai, Z.; Wang, Y.; Zhang, J.; Wu, H.; Wang, Y.; Li, C.; Li, D.; Lu, L.; Wang, X.; et al. Resveratrol ameliorates ionizing irradiation-induced long-term hematopoietic stem cell injury in mice. Free Radic. Biol. Med. 2013, 54, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Rimmele, P.; Lofek-Czubek, S.; Ghaffari, S. Resveratrol increases the bone marrow hematopoietic stem and progenitor cell capacity. Am. J. Hematol. 2014, 89, E235–E238. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, D.O.; Byun, J.E.; Kim, W.S.; Kim, M.J.; Song, H.Y.; Kim, Y.K.; Kang, D.K.; Park, Y.J.; Kim, T.D.; et al. Thioredoxin-interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat. Commun. 2016, 7, 13674. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Shin, J.Y.; Liu, Y.F.; Brown, K.; Luo, H.Z.; Xi, Y.N.; Haynes, C.M.; Chen, D. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Yokota, T.; Sudo, T.; Kondo, M.; Lai, A.; Kincade, P.W.; Kouro, T.; Iida, R.; Kokame, K.; Miyata, T.; et al. The Satb1 Protein Directs Hematopoietic Stem Cell Differentiation toward Lymphoid Lineages. Immunity 2013, 38, 1105–1115. [Google Scholar] [CrossRef]
- Todd, M.A.M.; Picketts, D.J. PHF6 Interacts with the Nucleosome Remodeling and Deacetylation (NuRD) Complex. J. Proteome Res. 2012, 11, 4326–4337. [Google Scholar] [CrossRef]
- Wendorff, A.A.; Aidan Quinn, S.; Alvarez, S.; Brown, J.A.; Biswas, M.; Gunning, T.; Palomero, T.; Ferrando, A.A. Epigenetic reversal of hematopoietic stem cell aging in Phf6-knockout mice. Nat. Aging 2022, 2, 1008–1023. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Erlandsson, E.; Kristiansen, T.; Lu, R.; Brakebusch, C.; Weissman, I.L.; Yuan, J.; Martin-Gonzalez, J.; Bryder, D. Clonal reversal of ageing-associated stem cell lineage bias via a pluripotent intermediate. Nat. Commun. 2017, 8, 14533. [Google Scholar] [CrossRef]
- Yang, J.H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305. [Google Scholar] [CrossRef]

{kind=link}
| Small Molecule | Targets | Functions |
|---|---|---|
| CASIN | Cdc42, elevate H4K16Ac | rejuvenate old HSCs, enhance lymphoid output, and reduce myeloid lineage output |
| VPA | HDACs | promote retention of HSCs stemness and prevent stress-induced HSC exhaustion |
| rapamycin | mTOR | restore the self-renewal and hematopoiesis of HSCs |
| TN13, SB203580 | p38 MAPK | protect HSCs from ROS–p38 MAPK induced exhaustion, rejuvenate aged HSCs |
| ABT263 | BCL-2 and BCL-xL | deplete senescent HSCs, attenuate HSC myeloid skewing, and improve HSC fitness |
| metformin | AMPK; not fully understood | improves defective hematopoiesis and reduces DNA damage |
| resveratrol | oxidant, sirtuins, and AMPK | increases HSC capacity and ameliorates irradiation-induced HSC injury |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, B.; Yuan, Q.; Guryanova, O.A. Epigenetic Mechanisms in Hematologic Aging and Premalignant Conditions. Epigenomes 2023, 7, 32. https://doi.org/10.3390/epigenomes7040032
Yan B, Yuan Q, Guryanova OA. Epigenetic Mechanisms in Hematologic Aging and Premalignant Conditions. Epigenomes. 2023; 7(4):32. https://doi.org/10.3390/epigenomes7040032
Chicago/Turabian StyleYan, Bowen, Qingchen Yuan, and Olga A. Guryanova. 2023. "Epigenetic Mechanisms in Hematologic Aging and Premalignant Conditions" Epigenomes 7, no. 4: 32. https://doi.org/10.3390/epigenomes7040032
APA StyleYan, B., Yuan, Q., & Guryanova, O. A. (2023). Epigenetic Mechanisms in Hematologic Aging and Premalignant Conditions. Epigenomes, 7(4), 32. https://doi.org/10.3390/epigenomes7040032