Abstract
Epigenetics generally involves genetic control by factors other than our own DNA sequence. Recent research has focused on delineating the mechanisms of two major epigenetic phenomena: DNA methylation and histone modification. As epigenetics involves many cellular processes, it is no surprise that it can also influence disease-associated gene expression. A direct link between respiratory infections, host cell epigenetic regulations, and chronic lung diseases is still unknown. Recent studies have revealed bacterium- or virus-induced epigenetic changes in the host cells. In this review, we focused on respiratory pathogens (viruses, bacteria, and fungi) induced epigenetic modulations (DNA methylation and histone modification) that may contribute to lung disease pathophysiology by promoting host defense or allowing pathogen persistence.
Keywords:
virus; epigenetics; bacteria; fungi; histone modification; DNA methylation; RSV; transcriptional factors 1. Introduction
Pulmonary pathogens have been responsible for some of the biggest global health crises in humans for centuries. This is in part due to the efficiency of their mode of transmission. However, their ability to manipulate the human immune system has also been observed as a significant factor. It has already been known that epigenetic alterations play an important role in pathology as well as their ability to modify the immune response [1]. Epigenetic modifications associated with nutrition and the microbiome and their contribution to long-term health development have been well documented [2]. Importantly, the recent research successes show how pulmonary pathogens can modulate the human genome and cause chronic diseases due to the advancement in the OMICS sciences, such as genomics, transcriptomics, proteomics, or metabolomics. For example, genome-wide association studies have revealed genetic changes in different regions of the genome that are primarily associated with complex and nonmalignant respiratory diseases, e.g., chronic obstructive pulmonary disease (COPD), asthma, and pulmonary arterial hypertension (PAH) [3,4,5]. Benincasa et al., have reviewed the epigenetic role in COPD, asthma, and PAH [6]. The epigenetic markers for these common chronic respiratory diseases were identified, e.g., DNA methylation, histone modification, and microRNA (miRNA) [7,8,9,10]. There is growing evidence supporting bacterial infection-induced epigenetic modification in the host by different mechanisms [11]. Viruses are intracellular parasites that continuously utilize the subordination and exploitation of cellular machinery for transcription and translation. Thus, it is no surprise that viruses induce modulation of host chromatin dynamics and transcription regulation, and examples of modulated epigenetic mechanisms are DNA methylation, histone post-translational modification, and transcription modification [12,13]. Bacterial virulence factors can modulate host cell genetic expression rates, limiting the ability of the immune system to adequately respond and allowing replicating bacteria to evade phagocytosis. Multiple virulence factors have been identified in recent studies that may be able to cause lasting pathogenic changes to host cell genomes, some of which may be associated with chronic pathologies such as allergy development, asthma, latent infection, carcinogenesis, and COPD exacerbation. Previously described epigenetic genome modifications, including histone modification, miRNA processing, DNA methylation and acetylation, protein chaperones, and protein degradation processes, all have the potential to upregulate and downregulate host genes at the whim of the invading organism [1,14,15]. These epigenetic changes have potentially chronic or permanent consequences for the host, which can last long after the resolution of the initial infection due to the persistence of some of these changes throughout generations of proliferating cells. Linking the virulence factors of common respiratory infections to consistent host genome modifications and finally to measurable long-term pathogenic outcomes is a potential new front for pulmonary research. The field of infectious epigenetics is still in its early stages, but many authors have already extensively cataloged the most common pathogens and their respective epigenetic targets [1,14,15].
In this review, we have discussed pulmonary pathogens (viruses, bacteria, and fungi) induced epigenetic alterations and their role in typical cell pathology. We highlighted the current understanding of how host-bacterial, viral, or fungal infections contribute changes to the epigenetic landscape and whether those interactions are responsible for the downstream effects contributing to chronic pulmonary diseases.
2. Epigenetic Mechanisms Regulate Gene Expression
The cells of eukaryotes are genetically homogenous but phenotypically heterogenous due to the differential expression of genes. One of the ways of regulating differential gene expression is epigenetically, which does not need mutation but is heritable [16]. Pathogens (bacterium, virus, and fungus) are known to modulate diverse epigenetic factors, namely histone modification, DNA methylation, chromatin-associated complexes, non-coding RNAs, and RNA splicing factors [11]. Here, we highlighted two major epigenetic mechanisms of gene regulation that have been reported to be modulated by pathogens (Figure 1): histone modifications [17] and DNA methylation [18].
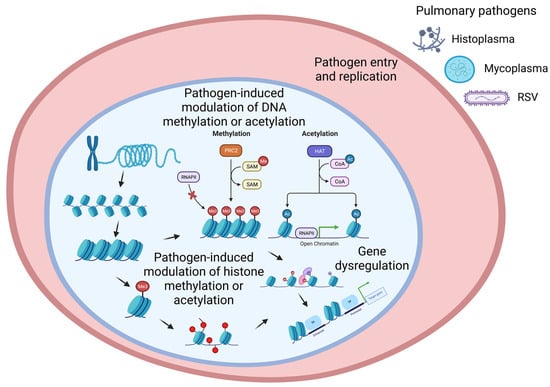
Figure 1.
A schematic of pulmonary pathogen-induced modulation of epigenetic factors. Pulmonary pathogens: bacterium (e.g., mycoplasma), virus (e.g., RSV), and fungus (e.g., histoplasma). Two major pathogen-driven epigenetic modulations of gene regulation: 1. DNA methylation or acetylation, and 2. Histone methylation or acetylation.
2.1. Histone Modification
Histone modification is the host cell’s ability to condense transcriptionally active euchromatin into highly condensed and silenced heterochromatin. The 147 base pairs of DNA wrapped around the histone octamers create a superstructure called a nucleosome that provides a highly ordered scaffold for condensing and storing DNA, and also uses steric hindrance to restrict the access of RNA polymerase enzymes to the incorporated DNA strand and any included genes [17]. The host cell can use writer proteins to covalently modify the N-terminal histone tails at more than 200 different sites with phosphorylation, acetylation, acylation, citrullination, methylation, or ubiquitination to change their properties of interaction with surrounding nucleosomes. The specific pattern of histone modification can either move nucleosome complexes along the strand of DNA, exposing particular genes for transcription, or create transcriptionally silent supercoils [19]. Each post-translational modification exerts electrochemical “cross-talk” on other covalent groups on the histone, creating a complex pattern of modification that can drastically influence the way a cell behaves. Proteins called erasers are responsible for removing these post-translational modifications, creating a dynamic balance between writing and erasing these groups that can be quickly modulated to respond to an infectious threat [20]. Multiple species of bacteria can secrete virulence factors that can directly remove the covalent modifications on the histone tails, modulating transcriptional activity for their own benefit [14,21]. Many virulence factors have the effect of slowing the immune response by suppressing transcription of proinflammatory proteins, thereby inhibiting immune cascades and the recruitment of additional immune cells [17]. Chlamydia trachomatis uses this mechanism by secreting a nuclear effector protein with histone N-lysine methyltransferase activity and histone demethylase activity to specifically modulate the methylation of histones H2B, H3, and H4. This subversion of host transcription can cause a global reduction in overall genetic activity and severely limit the cell’s ability to secrete alarm chemokines [22].
Another pathogenic histone modification mechanism can be accomplished through the modulation of histone-modifying enzymes, impairing the host’s ability to sense and respond to immune threats. Acetylation of histone tails at lysine residues creates a net negative charge, which can repel nearby nucleosomes and cause the chromatin to uncoil, but can have other varying effects based on other local post-translational modifications and their intrinsic cross-talk [23]. HDAC (Histone Deacetylase) proteins are responsible for removing acetyl groups when necessary, which often creates a heterochromatin state and silences genes [23]. This process has been shown to be upregulated during infections with Anaplasma phagocytophilum, Mycobacterium tuberculosis, and Pseudomonas aeruginosa, limiting the host cell’s ability to transcribe genes and respond to the infection. A well-defined example is the development of bacterial virulence factors that mimic the Ankyrin A protein and directly recruit and activate the HDAC1 enzymes at the site of transcriptionally active euchromatin. This causes localized hypoacetylation and gene suppression at the CYBB gene, which is essential for superoxide anion production and the proper function of the NADPH oxidase reaction [24].
2.2. DNA Methylation
DNA methylation is an easy way for a host cell to functionally silence transcription of a gene without excising the genetic code completely. DNA methyltransferases can quickly add a methyl group to a gene of interest at a cytosine or adenosine residue, creating methyl-cytosine or methyl-adenosine [18]. These bulky chemical groups inhibit the attachment of transcription factors and downregulate the activity of RNA polymerase and subsequent gene expression. Methyl groups will virtually silence all transcription, and acetyl groups tend to increase RNA polymerase activity at the site in question. These changes are persistent throughout generations of progenitor cells, creating chronic consequences for the host. Recognizing deleterious genomic methylations in a host cell that were specifically caused by an infection is difficult, and because of this, few pathogens have been associated with epigenetic methylations. Host cell methylations are usually localized to CpG islands in the genome, areas rich in cytosine and guanine residues, which allow for the activity of DNA-methyltransferase. Non-CpG methylations are one of the few true markers of pathogenic methylation [25]. Perhaps one of the best-characterized examples of this effect has been demonstrated by Mycobacterium tuberculosis Rv2699 protein secretion within host macrophages [25,26]. Multiple studies have shown various de novo methylations associated with Rv2699 proteins in macrophages infected with M. tuberculosis, with a possible decrease in IL-6 expression in infected cells [14]. Current reviews have also highlighted many different temporary epigenetic mechanisms used by pathogens to enhance virulence [1,14]. These changes are not passed through host progenitor cell lineages, so they are unlikely to cause chronic disease after infection, but many of these mechanisms may lead to clinically significant effects on the host beyond what may have been directly caused by the pathogen. Many questions remain about the long-term potential consequences of these infections. The exact mechanisms for the oncogenic epigenetic impacts of some infections like Helicobacter pylori and Human papillomavirus have been extensively documented [27,28,29,30].
3. Bacterial Infection-Mediated Pulmonary Epigenetics
Bacterial infections causing epigenetic change within a host nasal epithelial cell genome have been newly credited for being a significant pathogenic contributor to future chronic disease [11,31]. Ebenezer et al., have shown Pseudomonous aeruginosa infection drives epigenetic regulation in the host. They used the P. aeruginosa mouse model to test how the infection regulates nuclear spingosine-1-phosphate kinase 2 (SPHK2) and spingosine-1-phosphaet (SP1) and modulates epigenetic regulation in ling injury in vivo [32]. An in vitro study has also shown pathogenic bacterial infection-induced epigenetic modulation, e.g., flagellin of Legionella pneumophila is known to modulate histone (H3) acetylation or phosphorylation in infected lung epithelial cells [11]. Importantly, respiratory tract commensal bacteria, e.g., Moraxella catarrhalis, were also found to modulate acetylation or phosphorylation of H3 via the induction of an inflammatory signaling cascade (Figure 1) [33,34]. Denzer et al. [14] and Rajeev et al. [1] have comprehensively reviewed current research on epigenetic changes caused by bacterial infections, including many demonstrated epigenetic mechanisms of modification. Dupont et al. [35] define epigenetics as a change in gene function that is mitotically and/or meiotically heritable and that does not entail a change in DNA sequence. This is a rather classical perspective and excludes extra-genomic modifications done to the cell that have drastic effects on phenotype yet are not passed through progenitors. Interestingly, intrinsic, acquired, or adaptive forms of antimicrobial resistance can be characterized by different genetic and epigenetic molecular pathways [36,37,38]. Many current reviews of epigenetics have now expanded that definition to include those changes that are not passed through DNA replication and instead only affect cells directly modified by genetically active virulence factors [39]. For the purposes of this review and studying the effect of these diseases on the development of chronic ailments, we focused on the heritable epigenetic changes shown by human epithelial cells after a bacterial infection (Table 1).

Table 1.
Common pulmonary diseases and their associated epigenetic impacts due to bacterial infections.
4. Viral Infection-Mediated Pulmonary Epigenetics
Respiratory viral infections can trigger chronic lung diseases [63,64,65]. Asthma is a common chronic disease affecting children in Western countries. Although the pathophysiological changes underlying asthma development are complex and unknown, respiratory syncytial virus (RSV) and human rhinovirus virus (HRV) have been recognized as major etiological factors in wheezing illness, with a significant correlation with asthma development during childhood [66,67]. A recent number of studies have shown that respiratory viral infection causes epigenetic changes, which refer to genetic alterations that affect gene expression without any mutational genetic changes [64]. Here, we focused on a few common respiratory virus-driven epigenetic changes (Table 2 and Table 3).
4.1. Adenovirus
Adenovirus is a double-stranded DNA virus that commonly causes respiratory infections in humans. It uses a lytic type of life cycle, which is important in its pathophysiology [68]. A common theme in adenovirus infection is the suppression of the expression of most host genes except a few that are involved in cell division [69]. These profound changes are believed to be caused in part by epigenetic changes. ChIP-sip and ChIP-seq-based data analyses suggested that Adenovirus infection was associated with the modulation of two major epigenetic regulations: the organization of certain histone modifications and transcription factors within infected cells. The adenovirus e1a protein appeared to affect the acetylation of H3K9 and H3K18 and certain transcriptional factors [70,71]. Adenovirus protein VII is expressed late in the infected cells and translocates into the nucleus, which allows it to associate with host chromatin; consequently, it alters the chromatin composition of the infected cells. [72,73]. Additionally, the Adenovirus e1A protein is known to localize to host chromatin, alter host histone modifications, and overhaul transcription in infected cells. e1A binds to three different locations on the host’s chromatin. The first site is a region occupied by multiple genes related to the immune response of the host, and e1A binding downregulates these genes [72]. The second site of binding for this protein involves genes involved in the cell cycle. These genes were observed to have been upregulated [68,72]. The genes at the third site of binding are mostly involved in specialization and growth, and these genes were found to have been downregulated. These epigenetic changes hinder the immune system’s ability to respond to an adenovirus infection [72].
4.2. RSV
RSV is a negative-sense, single-stranded RNA virus enveloped by a protein coat. Since RSV affects the epithelial cells of the respiratory tract, the epigenetic factors that may induce changes in these cells are of particular interest. Caixia et al., underlined how the epigenetic dysregulation of signaling pathways in epithelial cells of the respiratory tract is related to shortfalls in immunological functions [74]. This implies that, through the induction of epigenetic modification, the virus can offset the immune response against it. Some of the signaling pathways that RSV can impact are tyrosine kinase growth factor signaling, the hexosamine biosynthetic pathway, and the extracellular matrix secretory pathways [75]. The virus can do this by inducing chromatin remodeling, which involves an increase in the number of nucleosome-free regions. This remodeling affects genes that regulate the aforementioned pathways. The increased accessibility of specific chromatin regions directly affects TGFβ-ECM and HBP pathways, which may explain the airway remodeling that happens in RSV infection [75].
Fonseca et al. [76] discussed that the disparities in immune responses between adults and infants are caused by epigenetic alterations of inflammatory genes. Histone methylation caused by RSV infection has been seen to enhance the production of Th2 cytokines after diminishing the production of pro-inflammatory cytokines. Other observations that have been made in mouse models suggest that RSV is also able to induce changes in non-coding RNAs [76]. These changes were seen to have impacts after the infection had subsided, which involved allergic asthma [76]. By having these effects, the virus is able to increase its own virulence while also having lasting effects, especially on vulnerable hosts.
4.3. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
SARS-CoV-2 is a positive-sense, single-stranded RNA virus that was first identified in the city of Wuhan, China. It is the virus responsible for the respiratory disease coronavirus disease 2019 (COVID-19). The virus has been found to alter the epigenetic environment of the host cell to increase its virulence by disrupting the host’s ability to recognize and respond to the pathogen [77]. Arguably the most significant regulator of the severity of COVID-19 infection is correlated with the increased expression of the receptor ACE2. DNA methylation is one way in which this receptor can be altered by the virus. Decreased DNA methylation close to the transcription site was found to result in increased expression of ACE2 in lung epithelial cells [77]. Chlamydas et al., suggested that the upregulation of the ACE2 receptor on the host cells by the virus is through histone modifications at the DNA packaging histones H3, H3K4me1, H3K4me3, and H3K27Ac [77]. Ozturkler et al., summarized how epigenetic changes such as hypermethylation of IFN genes and hypomethylation of inflammatory genes can be induced by the virus, and this change can be cell-specific [78]. Corley et al., described how, in severe cases of COVID-19, the epigenetic changes in immune cells varied and DNA methylation decreased in primary neutrophils [79].
Table 2.
Common viral pulmonary pathogens and their associated epigenetic changes.
Table 2.
Common viral pulmonary pathogens and their associated epigenetic changes.
| Pulmonary Pathogen | Epigenetic Changes | Cellular Impact | References |
|---|---|---|---|
| Adenovirus | Histone deacetylase inhibitor suppresses host HDAC proteins | Increased global acetylation, transcription modulation | [68,72] |
| Respiratory syncytial virus (RSV) | Chromatin modifications inducing nucleosome-free regions Demethylation of Nodal promoter Increased TGFβ expression | Impacts tyrosine kinase growth factor signaling, affects extracellular matrix secretory pathways Reduced pro-inflammatory cytokines release | [74,75,76] |
| Severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) | Hypermethylation of IFN related Hypomethylation of inflammatory genes Perturbation of epigenetic clock IRF1 and IRF7 were upregulated Epi-factors HDAC9 and SIRT1 were deregulated. | Increased level of ACE2 receptor expression Increased cytokine release | [77,78,79,80] |
| Influenza virus | Post translational histone modification Decrease in histone acetylation Demethylation on CREB1 binding region | Hypercytokinemia | [81,82,83] |
| Rhino virus | Modifications in DNA methylation | [64] | |
| Middle East respiratory syndrome-related corona virus (MERS) | H3K27 methylation | Downregulate antigen-presenting molecules | [13,77,83] |
TGFβ: tumor growth factor β; IFN: interferon; IFR1: interferon regulatory factor 1; IRF7: interferon regulatory factor 7; SIRT1: sirtuin 1; ACE2: angiotensin converting enzyme 2; CREB1: CAMP responsive element binding protein 1; H3K27: histone 3 at lysine residue in position 27.
Table 3.
Main types of epigenetic changes and virus presence in common respiratory viral diseases.
Table 3.
Main types of epigenetic changes and virus presence in common respiratory viral diseases.
| Types of Epigenetic Change | Adenovirus | SARS-CoV-2 | RSV | Influenza Virus | Rhino Virus | MERS |
|---|---|---|---|---|---|---|
| Chromatin remodeling | Not Identified | Not Identified | Yes [75] | Yes [83] | Not Identified | Not Identified |
| Changes in DNA methylation | Yes [68] | Yes [77,78,79] | Yes [74] | Yes [81,82] | Yes [64] | Yes [83] |
| Non-coding RNA | Yes [80] | Yes [74] | ||||
| Changes in DNA acetylation | Yes [68] | Not Identified | Not Identified | Yes [81] | Not Identified | Not Identified |
5. Fungal Epigenetics
Human pathogenic fungi-induced epigenetic changes are not well studied like viruses and bacteria, although there is a plethora of research into plant pathogenic fungi [84]. One cause for this lack of human-centered research could be the population affected by pathogenic fungi. Most fungal infections, including fungal respiratory infections, are asymptomatic in healthy populations [85]. Patients with asymptomatic fungal infections are less likely to seek medical care and thus pose no additional burden on the healthcare system. However, when pathogenic fungi infect vulnerable or immunocompromised patients, the infections may become invasive and require hospitalization [86,87,88]. A smaller patient population that is afflicted with fungal infections may be less enticing to researchers as compared with the vast population that can be infected with a bacterium like Streptococcus pneumoniae [88,89].
However, despite the smaller proportion of patients affected by fungal disease, the burden of fungal infections on the United States healthcare system should not be understated. A recent study from the Open Forum on Infectious Diseases, a publication from the Infectious Diseases Society of America, shows that fungal infections cost the United States approximately $6.7 billion in 2018. This study attributes 76.3% of the fungal infections and 81.1% of the associated costs to three pathogens: Aspergillus, Pneumocystis, and Candida [90]. All three of those fungi cause severe pulmonary disease in immunocompromised patients.
Fungal infections can impact the treatment and care of patients who are both immunocompromised and immunocompetent. In patients with chronic conditions like COPD or asthma, additional fungal infections complicate their care and may cause a severe exacerbation that requires hospitalization. A key factor contributing to all three chronic diseases (asthma, COPD, and cystic fibrosis) is mucus hypersecretion. Although epigenetic regulation of mucus hypersecretion remains unknown, a comprehensive review of epigenetic research on the mucus hypersecretion aspect of these chronic diseases reveals genes associated with DNA methylation and histone modification are major contributing factors [91]. One indication of fungal infection-induced epigenetic changes could be extrapolated from the funding by Perez FJ et al. They showed that fungal colonization with Pneumocystis has been shown to elevate the concentration of chloride channel accessory 1 (hCLCA1) in the infant lung parenchyma, and it also correlates with overproduction of mucin 5 AC (MUC5AC) [92]. Co-infections of fungal disease and viral infections, such as a co-infection of Aspergillus and SARS-CoV-2, can complicate the treatment of both diseases and have been attributed to poorer patient outcomes [93].
6. Conclusions
Genome-wide studies for characterization of epigenetic changes during pulmonary pathogen infection provide a better understanding of the complex relationship between pathogen infections and pulmonary diseases and even chronic lung diseases, e.g., asthma and COPD. However, we need to emphasize whether epigenetic changes directly correlate with changes in biological functions. Less common pathogens have still not been fully studied in the context of epigenetic modulations, and notably, neither have the long-term clinical consequences of these infections. Identifying which pathogens pose the greatest risk for the development of chronic disease and delineating the mechanism of their epigenetic changes can help focus public health efforts on the prevention or elimination of those diseases.
Author Contributions
Conceptualization, D.W., M.B., Y.A. and M.M. writing—review and editing, D.W., M.B., Y.A. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by NIH P20GM113123 and a pilot grant from UND SMHS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are thankful to Muhammad Faheem for his editing help with the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Rajeev, R.; Dwivedi, A.P.; Sinha, A.; Agarwaal, V.; Dev, R.R.; Kar, A.; Khosla, S. Epigenetic interaction of microbes with their mammalian hosts. J. Biosci. 2021, 46, 94. [Google Scholar] [CrossRef] [PubMed]
- Indrio, F.; Martini, S.; Francavilla, R.; Corvaglia, L.; Cristofori, F.; Mastrolia, S.A.; Neu, J.; Rautava, S.; Russo Spena, G.; Raimondi, F.; et al. Epigenetic Matters: The Link between Early Nutrition, Microbiome, and Long-term Health Development. Front. Pediatr. 2017, 5, 178. [Google Scholar] [CrossRef] [PubMed]
- Maselli, D.J.; Bhatt, S.P.; Anzueto, A.; Bowler, R.P.; DeMeo, D.L.; Diaz, A.A.; Dransfield, M.T.; Fawzy, A.; Foreman, M.G.; Hanania, N.A.; et al. Clinical Epidemiology of COPD: Insights From 10 Years of the COPDGene Study. Chest 2019, 156, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Batai, K.; Bleda, M.; Haimel, M.; Southgate, L.; Germain, M.; Pauciulo, M.W.; Hadinnapola, C.; Aman, J.; Girerd, B.; et al. Genetic determinants of risk in pulmonary arterial hypertension: International genome-wide association studies and meta-analysis. Lancet Respir. Med. 2019, 7, 227–238. [Google Scholar] [CrossRef]
- Sakornsakolpat, P.; Prokopenko, D.; Lamontagne, M.; Reeve, N.F.; Guyatt, A.L.; Jackson, V.E.; Shrine, N.; Qiao, D.; Bartz, T.M.; Kim, D.K.; et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat. Genet. 2019, 51, 494–505. [Google Scholar] [CrossRef]
- Benincasa, G.; DeMeo, D.L.; Glass, K.; Silverman, E.K.; Napoli, C. Epigenetics and pulmonary diseases in the horizon of precision medicine: A review. Eur. Respir. J. 2021, 57, 2003406. [Google Scholar] [CrossRef]
- Hoang, T.T.; Sikdar, S.; Xu, C.J.; Lee, M.K.; Cardwell, J.; Forno, E.; Imboden, M.; Jeong, A.; Madore, A.M.; Qi, C.; et al. Epigenome-wide association study of DNA methylation and adult asthma in the Agricultural Lung Health Study. Eur. Respir. J. 2020, 56, 2000217. [Google Scholar] [CrossRef]
- Napoli, C.; Benincasa, G.; Loscalzo, J. Epigenetic Inheritance Underlying Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 653–664. [Google Scholar] [CrossRef]
- Regan, E.A.; Hersh, C.P.; Castaldi, P.J.; DeMeo, D.L.; Silverman, E.K.; Crapo, J.D.; Bowler, R.P. Omics and the Search for Blood Biomarkers in Chronic Obstructive Pulmonary Disease. Insights from COPDGene. Am. J. Respir. Cell Mol. Biol. 2019, 61, 143–149. [Google Scholar] [CrossRef]
- DeVries, A.; Vercelli, D. Epigenetic Mechanisms in Asthma. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S1), S48–S50. [Google Scholar] [CrossRef]
- Bierne, H.; Hamon, M.; Cossart, P. Epigenetics and bacterial infections. Cold Spring Harb. Perspect. Med. 2012, 2, a010272. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.; Budd, A.; Bielawski, J.P. Introduction to Genome Biology and Diversity. Methods Mol. Biol. 2019, 1910, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Albarran, M.; Navarro-Delgado, E.I.; Del Moral-Morales, A.; Alcaraz, N.; Baumbach, J.; Gonzalez-Barrios, R.; Soto-Reyes, E. Comparative transcriptome analysis reveals key epigenetic targets in SARS-CoV-2 infection. NPJ Syst. Biol. Appl. 2021, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Denzer, L.; Schroten, H.; Schwerk, C. From gene to protein—How bacterial virulence factors manipulate host gene expression during infection. Int. J. Mol. Sci. 2020, 21, 3730. [Google Scholar] [CrossRef] [PubMed]
- De Monerri, N.C.S.; Kim, K. Pathogens hijack the epigenome: A new twist on host-pathogen interactions. Am. J. Pathol. 2014, 184, 897–911. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33 (Suppl. S3), 245–254. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Niller, H.H.; Masa, R.; Venkei, A.; Mészáros, S.; Minarovits, J. Pathogenic mechanisms of intracellular bacteria. Curr. Opin. Infect. Dis. 2017, 30, 309–315. [Google Scholar] [CrossRef]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef]
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes 2021, 12, 1596. [Google Scholar] [CrossRef]
- Hamon, M.A.; Batsché, E.; Régnault, B.; Tham, T.N.; Seveau, S.; Muchardt, C.; Cossart, P. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. USA 2007, 104, 13467–13472. [Google Scholar] [CrossRef]
- Pennini, M.E.; Perrinet, S.; Dautry-Varsat, A.; Subtil, A. Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis. PLoS Pathog. 2010, 6, e1000995. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Rennoll-Bankert, K.E.; Garcia-Garcia, J.C.; Sinclair, S.H.; Dumler, J.S. Chromatin-bound bacterial effector ankyrin A recruits histone deacetylase 1 and modifies host gene expression. Cell. Microbiol. 2015, 17, 1640–1652. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Upadhyay, S.; Srilalitha, M.; Nandicoori, V.K.; Khosla, S. The interaction of mycobacterial protein Rv2966c with host chromatin is mediated through non-CpG methylation and histone H3/H4 binding. Nucleic Acids Res. 2015, 43, 3922–3937. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Sowpati, D.T.; Singh, P.; Khan, M.Z.; Ganji, R.; Upadhyay, S.; Banerjee, S.; Nandicoori, V.K.; Khosla, S. Genome-wide non-CpG methylation of the host genome during M. tuberculosis infection. Sci. Rep. 2016, 6, 25006. [Google Scholar] [CrossRef]
- Ding, S.-Z.; Fischer, W.; Kaparakis-Liaskos, M.; Liechti, G.; Merrell, D.S.; Grant, P.A.; Ferrero, R.L.; Crowe, S.E.; Haas, R.; Hatakeyama, M. Helicobacter pylori-induced histone modification, associated gene expression in gastric epithelial cells, and its implication in pathogenesis. PLoS ONE 2010, 5, e9875. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Compare, D.; De Colibus, P.; De Nucci, G.; Rocco, A. Helicobacter pylori and epigenetic mechanisms underlying gastric carcinogenesis. Dig. Dis. 2007, 25, 225–229. [Google Scholar] [CrossRef]
- Santos, J.C.; Ribeiro, M.L. Epigenetic regulation of DNA repair machinery in Helicobacter pylori-induced gastric carcinogenesis. World J. Gastroenterol. WJG 2015, 21, 9021. [Google Scholar] [CrossRef] [PubMed]
- Soto, D.; Song, C.; McLaughlin-Drubin, M.E. Epigenetic alterations in human papillomavirus-associated cancers. Viruses 2017, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Al Akeel, R. Role of epigenetic reprogramming of host genes in bacterial pathogenesis. Saudi J. Biol. Sci. 2013, 20, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Berdyshev, E.V.; Bronova, I.A.; Liu, Y.; Tiruppathi, C.; Komarova, Y.; Benevolenskaya, E.V.; Suryadevara, V.; Ha, A.W.; Harijith, A.; et al. Pseudomonas aeruginosa stimulates nuclear sphingosine-1-phosphate generation and epigenetic regulation of lung inflammatory injury. Thorax 2019, 74, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.; Holt, L.; Kim, S.C.; Schwabe, R.F.; Sartor, R.B.; Jobin, C. Transforming growth factor-beta 1 inhibits non-pathogenic Gram negative bacteria-induced NF-kappa B recruitment to the interleukin-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J. Biol. Chem. 2003, 278, 23851–23860. [Google Scholar] [CrossRef] [PubMed]
- Slevogt, H.; Schmeck, B.; Jonatat, C.; Zahlten, J.; Beermann, W.; van Laak, V.; Opitz, B.; Dietel, S.; N’Guessan, P.D.; Hippenstiel, S.; et al. Moraxella catarrhalis induces inflammatory response of bronchial epithelial cells via MAPK and NF-κB activation and histone deacetylase activity reduction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L818–L826. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. In Seminars in Reproductive Medicine; Thieme Medical Publishers: New York, NY, USA, 2009. [Google Scholar]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Motta, S.S.; Cluzel, P.; Aldana, M. Adaptive resistance in bacteria requires epigenetic inheritance, genetic noise, and cost of efflux pumps. PLoS ONE 2015, 10, e0118464. [Google Scholar] [CrossRef]
- Cohen, N.R.; Lobritz, M.A.; Collins, J.J. Microbial persistence and the road to drug resistance. Cell Host Microbe 2013, 13, 632–642. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Varghese, T.; Jayasri, M.; Suthindhiran, K. Marine A ctinomycetes as potential source for histone deacetylase inhibitors and epigenetic modulation. Lett. Appl. Microbiol. 2015, 61, 69–76. [Google Scholar] [CrossRef]
- Li, T.; Lu, Q.; Wang, G.; Xu, H.; Huang, H.; Cai, T.; Kan, B.; Ge, J.; Shao, F. SET-domain bacterial effectors target heterochromatin protein 1 to activate host rDNA transcription. EMBO Rep. 2013, 14, 733–740. [Google Scholar] [CrossRef]
- Krishnananthasivam, S.; Jayathilaka, N.; Sathkumara, H.D.; Corea, E.; Natesan, M.; De Silva, A.D. Host gene expression analysis in Sri Lankan melioidosis patients. PLoS Neglected Trop. Dis. 2017, 11, e0005643. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Curry, H.M.; Zwilling, B.S.; Lafuse, W.P. Mycobacteria inhibition of IFN-γ induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J. Immunol. 2005, 174, 5687–5694. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, I.; Kaur, P.; Nandicoori, V.K.; Khosla, S. Mycobacteria modulate host epigenetic machinery by Rv1988 methylation of a non-tail arginine of histone H3. Nat. Commun. 2015, 6, 8922. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Yi, M.; Chen, J.; Li, S.; Chen, W. Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3. Biochem. Biophys. Res. Commun. 2016, 473, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Jose, L.; Ramachandran, R.; Bhagavat, R.; Gomez, R.L.; Chandran, A.; Raghunandanan, S.; Omkumar, R.V.; Chandra, N.; Mundayoor, S.; Kumar, R.A. Hypothetical protein Rv3423. 1 of Mycobacterium tuberculosis is a histone acetyltransferase. FEBS J. 2016, 283, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Komar, D.; Juszczynski, P. Rebelled epigenome: Histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin. Epigenet. 2020, 12, 147. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Feng, C.; Zhai, Y.Z.; Zhou, X.; Li, B.; Wang, L.L.; Chen, W.; Lv, F.Q.; Li, T.S. Identification of miRNA biomarkers of pneumonia using RNA-sequencing and bioinformatics analysis. Exp. Ther. Med. 2017, 13, 1235–1244. [Google Scholar] [CrossRef]
- Li, H.; Xu, H.; Zhou, Y.; Zhang, J.; Long, C.; Li, S.; Chen, S.; Zhou, J.-M.; Shao, F. The phosphothreonine lyase activity of a bacterial type III effector family. Science 2007, 315, 1000–1003. [Google Scholar] [CrossRef]
- Rolando, M.; Sanulli, S.; Rusniok, C.; Gomez-Valero, L.; Bertholet, C.; Sahr, T.; Margueron, R.; Buchrieser, C. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 2013, 13, 395–405. [Google Scholar] [CrossRef]
- Schuelein, R.; Spencer, H.; Dagley, L.F.; Li, P.F.; Luo, L.; Stow, J.L.; Abraham, G.; Naderer, T.; Gomez-Valero, L.; Buchrieser, C. Targeting of RNA Polymerase II by a nuclear Legionella pneumophila Dot/Icm effector SnpL. Cell. Microbiol. 2018, 20, e12852. [Google Scholar] [CrossRef]
- Von Dwingelo, J.; Chung, I.Y.W.; Price, C.T.; Li, L.; Jones, S.; Cygler, M.; Abu Kwaik, Y. Interaction of the Ankyrin H core effector of legionella with the host LARP7 component of the 7SK snRNP complex. mBio 2019, 10, e01942-19. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; Winer, B.Y.; Jaganathan, A.; Patel, J.; Sgobba, M.; Schuch, R.; Gupta, Y.K.; Haider, S.; Wang, R.; Fischetti, V.A. Anthrax SET protein: A potential virulence determinant that epigenetically represses NF-κB activation in infected macrophages. J. Biol. Chem. 2013, 288, 23458–23472. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B.; Ravaux, L.; Mémet, S.; Wu, Y.; Sturny-Leclère, A.; Leduc, D.; Denoyelle, C.; Goossens, P.L.; Payá, M.; Raymondjean, M. Anthrax lethal toxin down-regulates type-IIA secreted phospholipase A2 expression through MAPK/NF-κB inactivation. Biochem. Pharmacol. 2010, 79, 1149–1155. [Google Scholar] [CrossRef]
- Ha, S.-D.; Reid, C.; Meshkibaf, S.; Kim, S.O. Inhibition of interleukin 1β (IL-1β) expression by anthrax lethal toxin (LeTx) is reversed by histone deacetylase 8 (HDAC8) inhibition in murine macrophages. J. Biol. Chem. 2016, 291, 8745–8755. [Google Scholar] [CrossRef]
- Murata, M.; Azuma, Y.; Miura, K.; Rahman, M.A.; Matsutani, M.; Aoyama, M.; Suzuki, H.; Sugi, K.; Shirai, M. Chlamydial SET domain protein functions as a histone methyltransferase. Microbiology 2007, 153, 585–592. [Google Scholar] [CrossRef]
- Mojica, S.A.; Hovis, K.M.; Frieman, M.B.; Tran, B.; Hsia, R.-c.; Ravel, J.; Jenkins-Houk, C.; Wilson, K.L.; Bavoil, P.M. SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol. Biol. Cell 2015, 26, 1918–1934. [Google Scholar] [CrossRef]
- Choung, H.-K.; Kim, Y.A.; Lee, M.J.; Kim, N.; Khwarg, S.I. Multigene methylation analysis of ocular adnexal MALT lymphoma and their relationship to Chlamydophila psittaci infection and clinical characteristics in South Korea. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Vdovikova, S.; Gilfillan, S.; Wang, S.; Dongre, M.; Wai, S.N.; Hurtado, A. Modulation of gene transcription and epigenetics of colon carcinoma cells by bacterial membrane vesicles. Sci. Rep. 2018, 8, 7434. [Google Scholar] [CrossRef]
- Bandyopadhaya, A.; Tsurumi, A.; Maura, D.; Jeffrey, K.L.; Rahme, L.G. A quorum-sensing signal promotes host tolerance training through HDAC1-mediated epigenetic reprogramming. Nat. Microbiol. 2016, 1, 16174. [Google Scholar] [CrossRef]
- Dortet, L.; Lombardi, C.; Cretin, F.; Dessen, A.; Filloux, A. Pore-forming activity of the Pseudomonas aeruginosa type III secretion system translocon alters the host epigenome. Nat. Microbiol. 2018, 3, 378–386. [Google Scholar] [CrossRef]
- Zughaier, S.M.; Rouquette-Loughlin, C.E.; Shafer, W.M. Identification of a Neisseria gonorrhoeae histone deacetylase: Epigenetic impact on host gene expression. Pathogens 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Hartert, T.V. Evidence for a causal relationship between respiratory syncytial virus infection and asthma. Expert Rev. Anti-Infect. Ther. 2011, 9, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Pech, M.; Weckmann, M.; Konig, I.R.; Franke, A.; Heinsen, F.A.; Oliver, B.; Ricklefs, I.; Fuchs, O.; Rabe, K.; Hansen, G.; et al. Rhinovirus infections change DNA methylation and mRNA expression in children with asthma. PLoS ONE 2018, 13, e0205275. [Google Scholar] [CrossRef]
- Britto, C.J.; Brady, V.; Lee, S.; Dela Cruz, C.S. Respiratory Viral Infections in Chronic Lung Diseases. Clin. Chest Med. 2017, 38, 87–96. [Google Scholar] [CrossRef]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef]
- Kusel, M.M.; de Klerk, N.H.; Kebadze, T.; Vohma, V.; Holt, P.G.; Johnston, S.L.; Sly, P.D. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 2007, 119, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Milavetz, B.I.; Balakrishnan, L. Viral epigenetics. In Cancer Epigenetics; Humana Press: New York, NY, USA, 2015; pp. 569–596. [Google Scholar]
- Tooze, J.; Acheson, N. DNA Tumor Viruses; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1980. [Google Scholar]
- Ferrari, R.; Pellegrini, M.; Horwitz, G.A.; Xie, W.; Berk, A.J.; Kurdistani, S.K. Epigenetic reprogramming by adenovirus e1a. Science 2008, 321, 1086–1088. [Google Scholar] [CrossRef]
- Ferrari, R.; Su, T.; Li, B.; Bonora, G.; Oberai, A.; Chan, Y.; Sasidharan, R.; Berk, A.J.; Pellegrini, M.; Kurdistani, S.K. Reorganization of the host epigenome by a viral oncogene. Genome Res. 2012, 22, 1212–1221. [Google Scholar] [CrossRef]
- Lynch, K.L.; Gooding, L.R.; Garnett-Benson, C.; Ornelles, D.A.; Avgousti, D.C. Epigenetics and the dynamics of chromatin during adenovirus infections. FEBS Lett. 2019, 593, 3551–3570. [Google Scholar] [CrossRef]
- Avgousti, D.C.; Herrmann, C.; Kulej, K.; Pancholi, N.J.; Sekulic, N.; Petrescu, J.; Molden, R.C.; Blumenthal, D.; Paris, A.J.; Reyes, E.D. A core viral protein binds host nucleosomes to sequester immune danger signals. Nature 2016, 535, 173–177. [Google Scholar] [CrossRef]
- Caixia, L.; Yang, X.; Yurong, T.; Xiaoqun, Q. Involvement of epigenetic modification in epithelial immune responses during respiratory syncytial virus infection. Microb. Pathog. 2019, 130, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiao, D.; Mann, M.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus infection induces chromatin remodeling to activate growth factor and extracellular matrix secretion pathways. Viruses 2020, 12, 804. [Google Scholar] [CrossRef]
- Fonseca, W.; Lukacs, N.W.; Ptaschinski, C. Factors affecting the immunity to respiratory syncytial virus: From epigenetics to microbiome. Front. Immunol. 2018, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Chlamydas, S.; Papavassiliou, A.G.; Piperi, C. Epigenetic mechanisms regulating COVID-19 infection. Epigenetics 2021, 16, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Ozturkler, Z.; Kalkan, R. A New Perspective of COVID-19 Infection: An Epigenetics Point of View. Glob. Med. Genet. 2022, 9, 004–006. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.J.; Pang, A.P.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26. [Google Scholar] [CrossRef]
- Khan, M.A.-A.-K.; Sany, M.R.U.; Islam, M.S.; Islam, A.B.M.M.K. Epigenetic regulator miRNA pattern differences among SARS-CoV, SARS-CoV-2, and SARS-CoV-2 world-wide isolates delineated the mystery behind the epic pathogenicity and distinct clinical characteristics of pandemic COVID-19. Front. Genet. 2020, 11, 765. [Google Scholar] [CrossRef]
- Marcos-Villar, L.; Diaz-Colunga, J.; Sandoval, J.; Zamarreno, N.; Landeras-Bueno, S.; Esteller, M.; Falcon, A.; Nieto, A. Epigenetic control of influenza virus: Role of H3K79 methylation in interferon-induced antiviral response. Sci. Rep. 2018, 8, 1230. [Google Scholar] [CrossRef]
- Mukherjee, S.; Vipat, V.C.; Chakrabarti, A.K. Infection with influenza A viruses causes changes in promoter DNA methylation of inflammatory genes. Influenza Other Respir. Viruses 2013, 7, 979–986. [Google Scholar] [CrossRef]
- Menachery, V.D.; Schafer, A.; Burnum-Johnson, K.E.; Mitchell, H.D.; Eisfeld, A.J.; Walters, K.B.; Nicora, C.D.; Purvine, S.O.; Casey, C.P.; Monroe, M.E.; et al. MERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proc. Natl. Acad. Sci. USA 2018, 115, E1012–E1021. [Google Scholar] [CrossRef]
- Doehlemann, G.; Okmen, B.; Zhu, W.; Sharon, A. Plant Pathogenic Fungi. Microbiol. Spectr. 2017, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, G.; Meng, G. Pathogenic Fungal Infection in the Lung. Front. Immunol. 2019, 10, 1524. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, S.; Moretti, S.; Perruccio, K.; Fallarino, F.; Bozza, S.; Montagnoli, C.; Mosci, P.; Lipford, G.B.; Pitzurra, L.; Romani, L. TLRs govern neutrophil activity in aspergillosis. J. Immunol. 2004, 173, 7406–7415. [Google Scholar] [CrossRef] [PubMed]
- Jose, R.J.; Brown, J.S. Opportunistic and fungal infections of the lung. Medicine 2012, 40, 335–339. [Google Scholar] [CrossRef]
- José, R.J.; Periselneris, J.N.; Brown, J.S. Opportunistic bacterial, viral and fungal infections of the lung. Medicine 2020, 48, 366–372. [Google Scholar] [CrossRef]
- Henriques-Normark, B.; Tuomanen, E.I. The pneumococcus: Epidemiology, microbiology, and pathogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a010215. [Google Scholar] [CrossRef]
- Rayens, E.; Norris, K.A. Prevalence and healthcare burden of fungal infections in the United States, 2018. In Open Forum Infectious Diseases; Oxford University Press: New York, NY, USA, 2022; p. ofab593. [Google Scholar]
- Saco, T.V.; Breitzig, M.T.; Lockey, R.F.; Kolliputi, N. Epigenetics of mucus hypersecretion in chronic respiratory diseases. Am. J. Respir. Cell Mol. Biol. 2018, 58, 299–309. [Google Scholar] [CrossRef]
- Pérez, F.J.; Ponce, C.A.; Rojas, D.A.; Iturra, P.A.; Bustamante, R.I.; Gallo, M.; Hananias, K.; Vargas, S.L. Fungal colonization with Pneumocystis correlates to increasing chloride channel accessory 1 (hCLCA1) suggesting a pathway for up-regulation of airway mucus responses, in infant lungs. Results Immunol. 2014, 4, 58–61. [Google Scholar] [CrossRef]
- Hoenigl, M. Invasive Fungal Disease Complicating Coronavirus Disease 2019: When It Rains, It Spores; Oxford University Press: New York, NY, USA, 2021; pp. e1645–e1648. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).