
Epigenome-Wide Changes in the Cell Layers of the Vein Wall When Exposing the Venous Endothelium to Oscillatory Shear Stress
 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
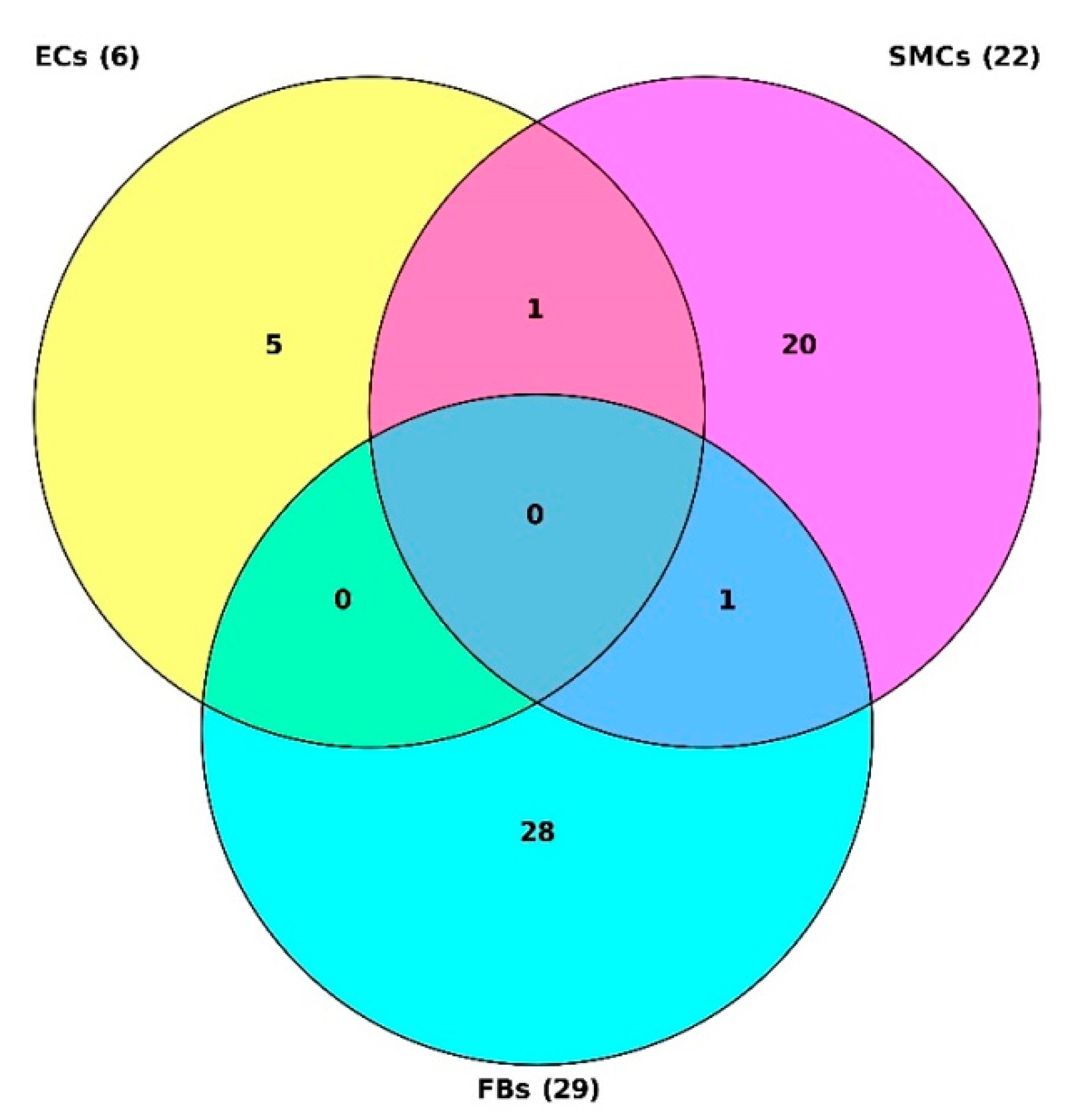
2.1. Identification of Target Genes
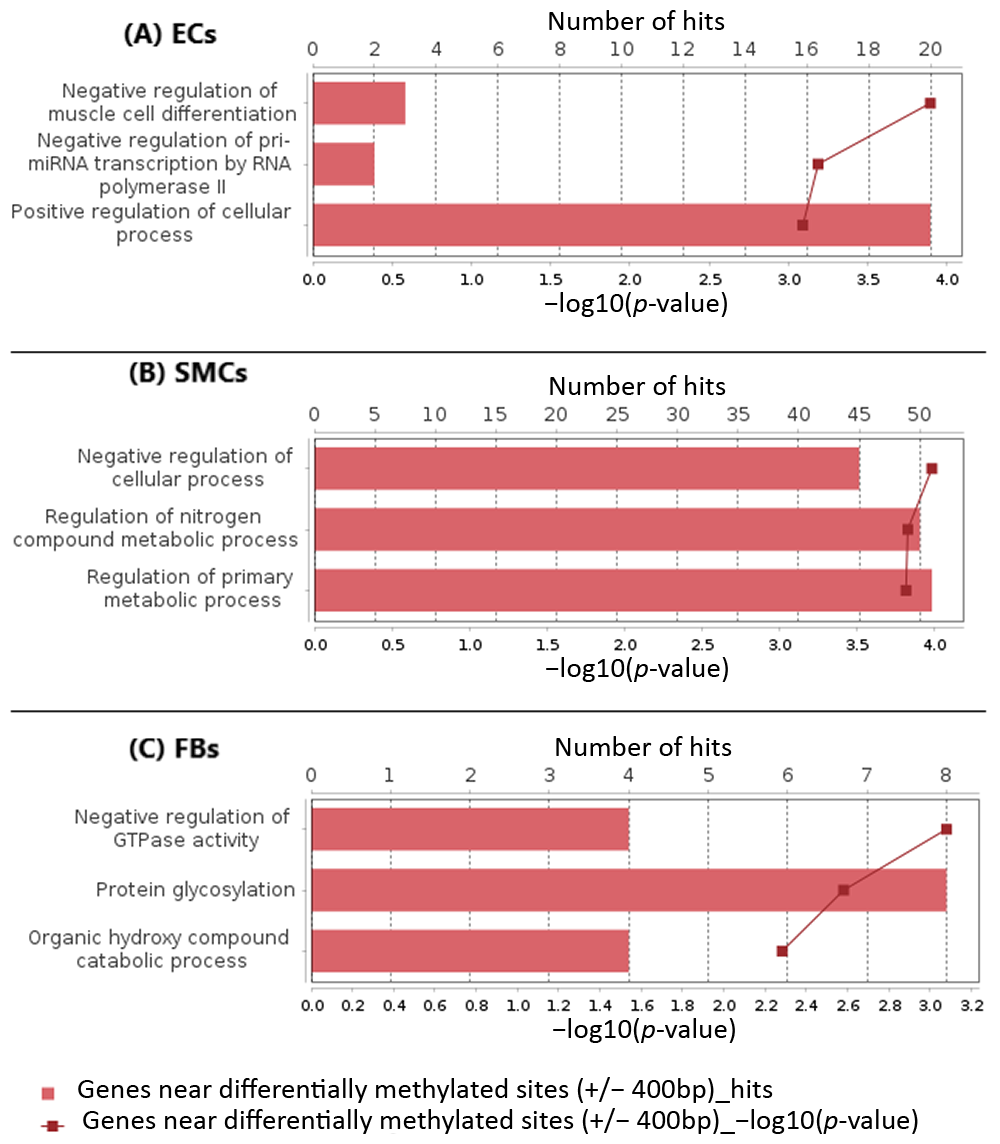
2.2. Functional Classification of Genes
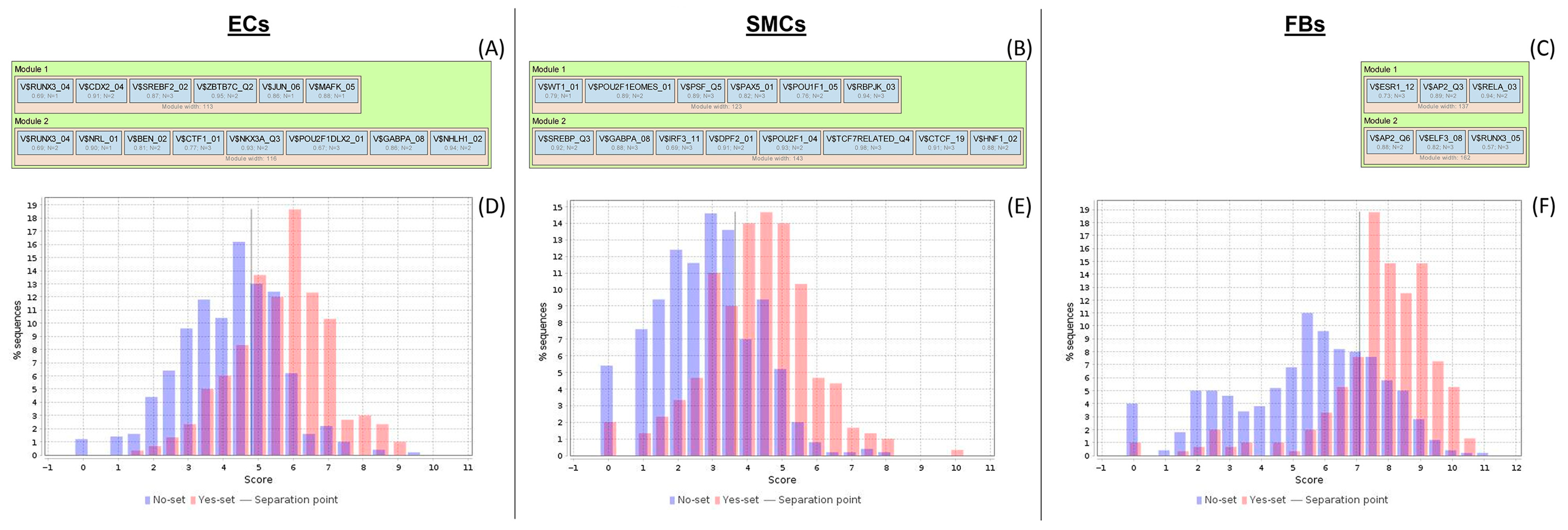
2.3. Analysis of Enriched Transcription Factor Binding Sites and Composite Modules
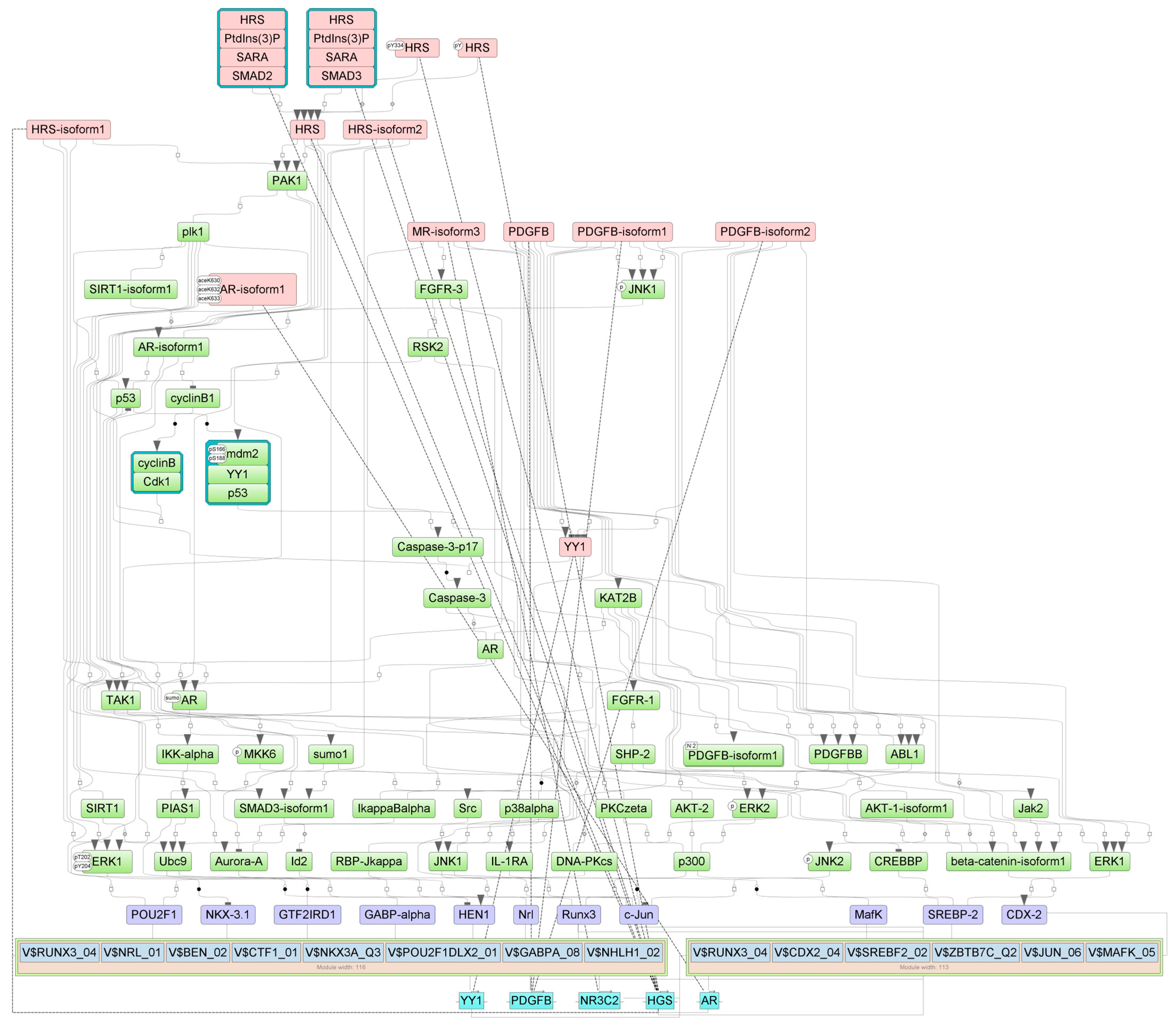
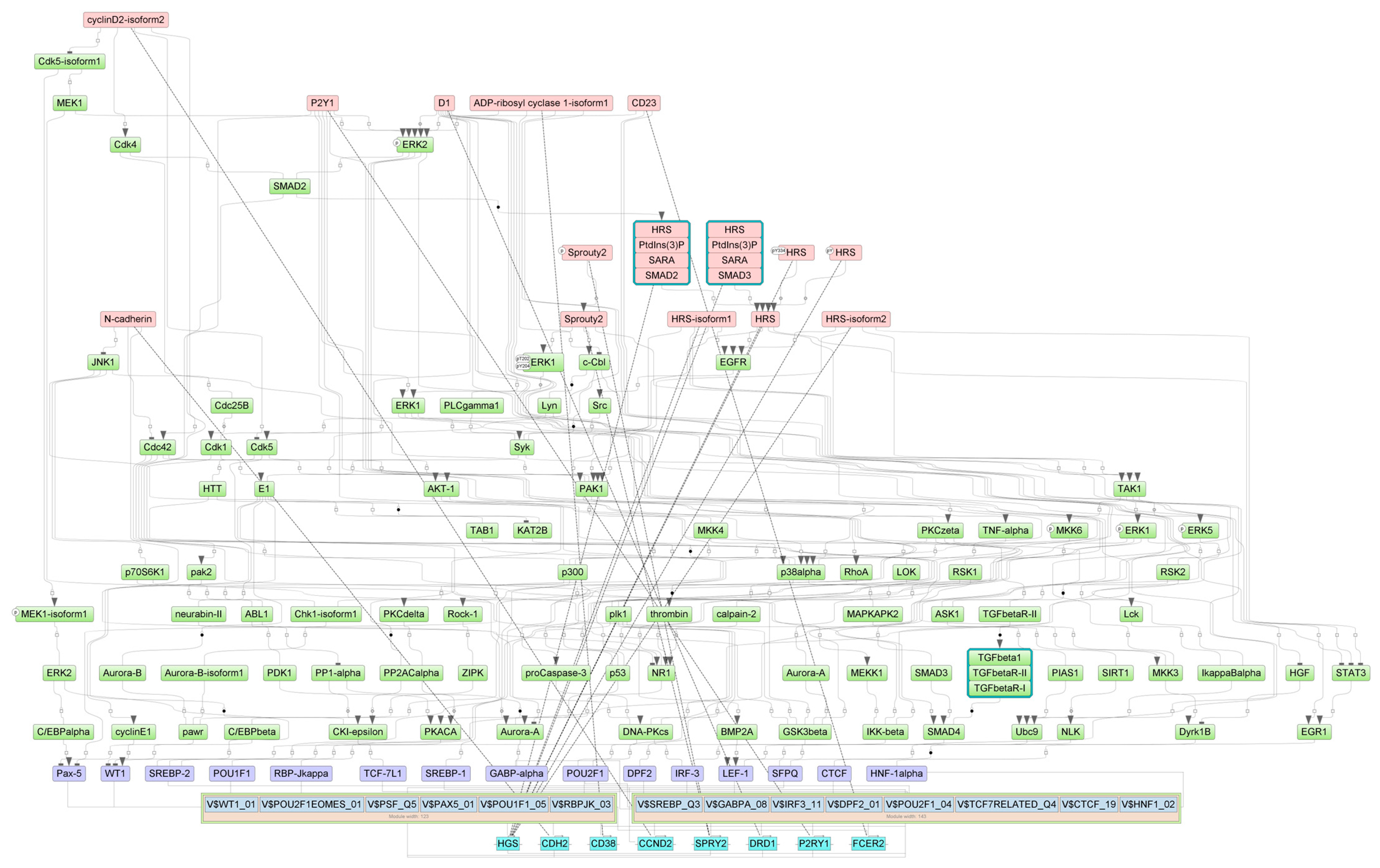
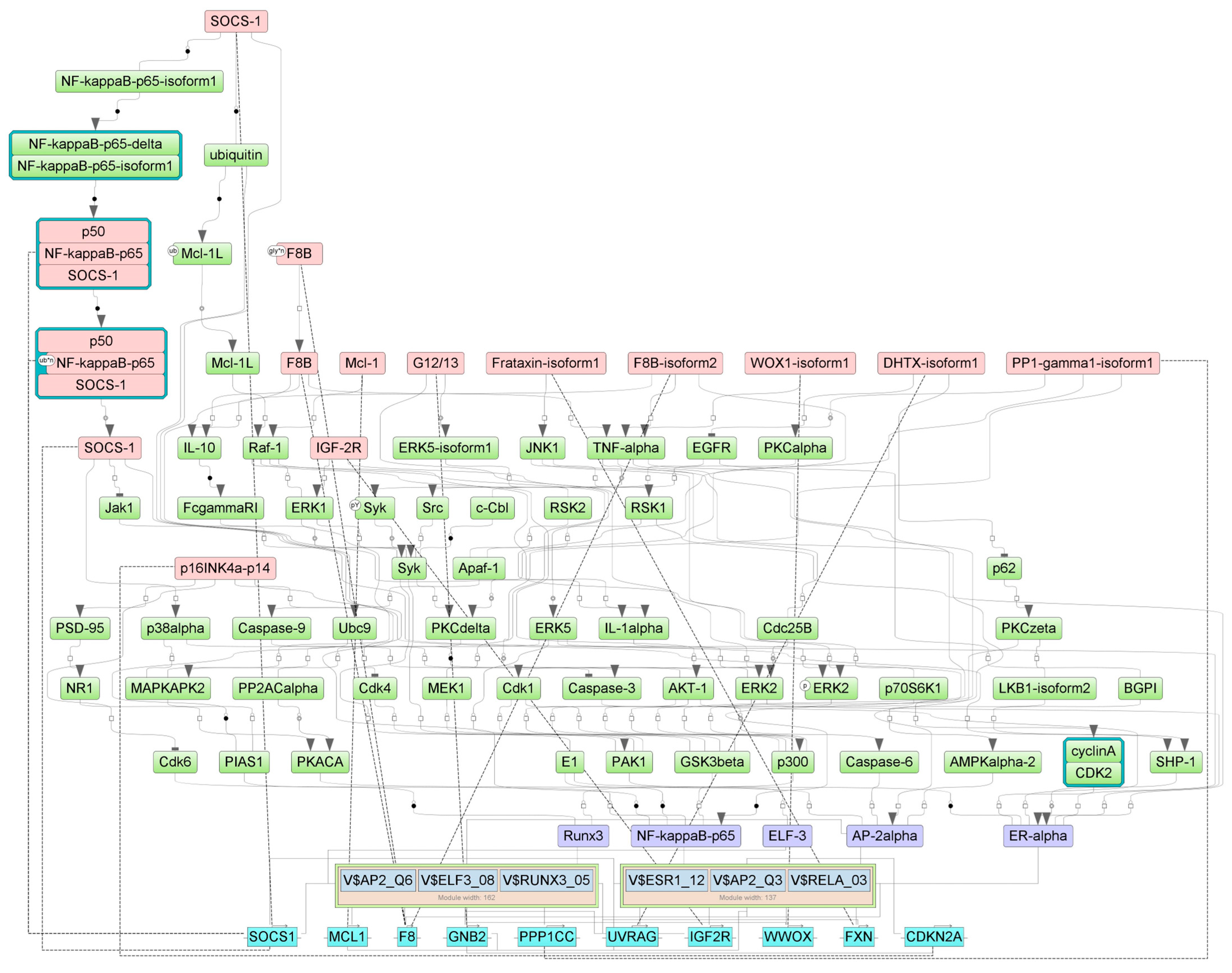
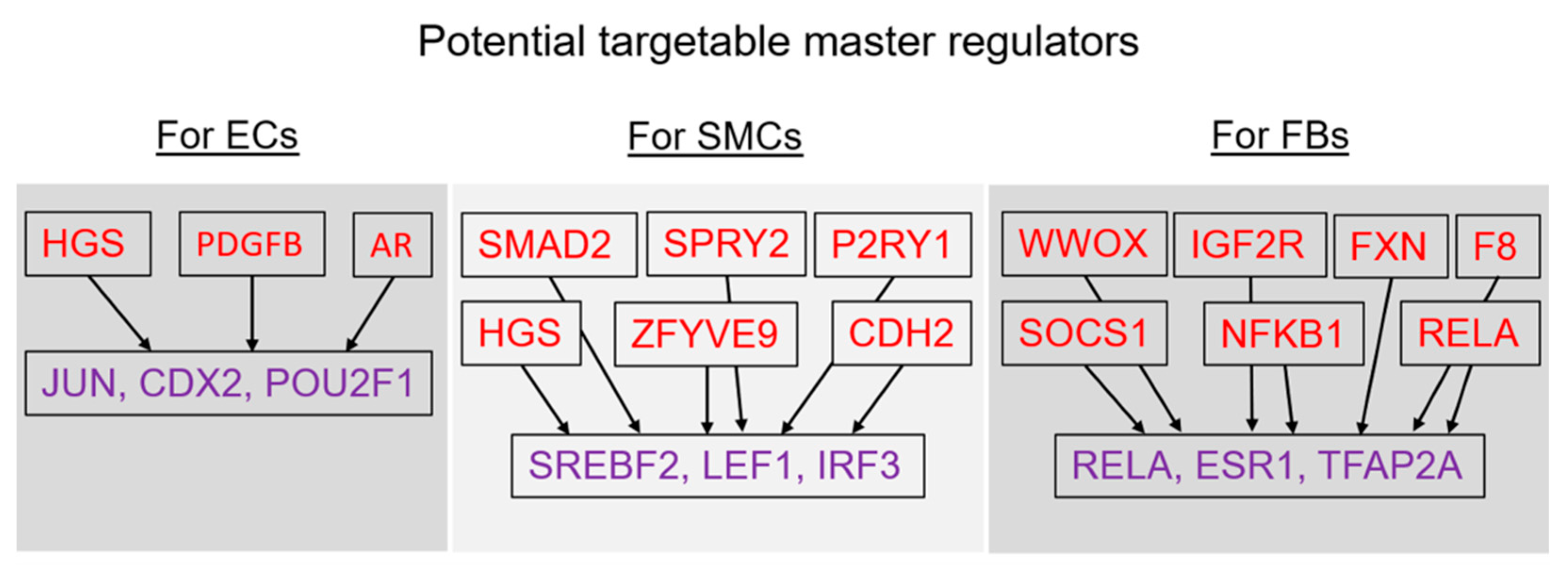
2.4. Finding Master Regulators in Networks
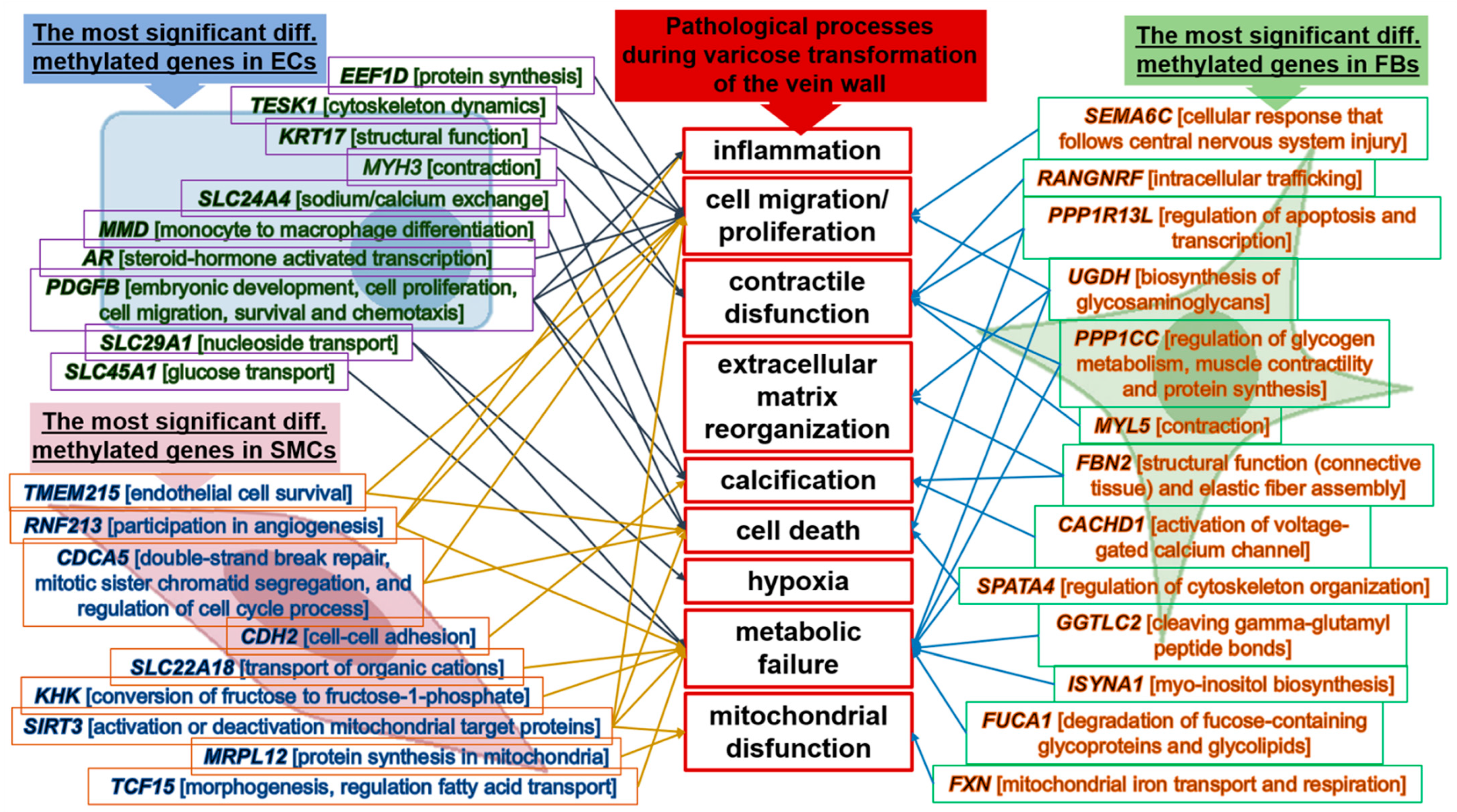
3. Discussion
4. Materials and Methods
4.1. Sample Preparation and Cell Culture Experiments
4.2. Epigenome-Wide DNA Methylation Analysis
4.3. Advanced Bioinformatics Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, M.L.; Bond, A.R.; George, S.J. Mechanobiology of the endothelium in vascular health and disease: In vitro shear stress models. Cardiovasc. Drugs Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Oklu, R.; Habito, R.; Mayr, M.; Deipolyi, A.R.; Albadawi, H.; Hesketh, R.; Walker, T.G.; Linskey, K.R.; Long, C.A.; Wicky, S.; et al. Pathogenesis of varicose veins. J. Vasc. Interv. Radiol. 2012, 23, 33–39. [Google Scholar] [CrossRef]
- Kuo, C.T.; Veselits, M.L.; Barton, K.P.; Lu, M.M.; Clendenin, C.; Leiden, J.M. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 1997, 11, 2996–3006. [Google Scholar] [CrossRef]
- Wasserman, S.M.; Topper, J.N. Adaptation of the endothelium to fluid flow: In vitro analyses of gene expression and in vivo implications. Vasc. Med. 2004, 9, 35–45. [Google Scholar] [CrossRef]
- Roux, E.; Bougaran, P.; Dufourcq, P.; Couffinhal, T. Fluid Shear Stress Sensing by the Endothelial Layer. Front. Physiol. 2020, 1, 861. [Google Scholar] [CrossRef]
- Urschel, K.; Tauchi, M.; Achenbach, S.; Dietel, B. Investigation of Wall Shear Stress in Cardiovascular Research and in Clinical Practice—From Bench to Bedside. Int. J. Mol. Sci. 2021, 22, 5635. [Google Scholar] [CrossRef]
- Dekker, R.J.; van Soest, S.; Fontijn, R.D.; Salamanca, S.; de Groot, P.G.; VanBavel, E.; Pannekoek, H.; Horrevoets, A.J.G. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002, 100, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Smetanina, M.A.; Kel, A.E.; Sevost’ianova, K.S.; Maiborodin, I.V.; Shevela, A.I.; Zolotukhin, I.A.; Stegmaier, P.; Filipenko, M.L. DNA methylation and gene expression profiling reveal MFAP5 as a regulatory driver of extracellular matrix remodeling in varicose vein disease. Epigenomics 2018, 10, 1103–1119. [Google Scholar] [CrossRef]
- Umehara, T. Epidrugs: Toward Understanding and Treating Diverse Diseases. Epigenomes 2022, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Smetanina, M.A.; Shevela, A.I.; Gavrilov, K.A.; Filipenko, M.L. The genetic constituent of varicose vein pathogenesis as a key for future treatment option development. Vessel Plus 2021, 5, 19. [Google Scholar] [CrossRef]
- Kel, A.; Voss, N.; Jauregui, R.; Kel-Margoulis, O.; Wingender, E. Beyond microarrays: Finding key transcription factors controlling signal transduction pathways. BMC Bioinform. 2006, 7, S13. [Google Scholar] [CrossRef]
- Stegmaier, P.; Voss, N.; Meier, T.; Kel, A.; Wingender, E.; Borlak, J. Advanced Computational Biology Methods Identify Molecular Switches for Malignancy in an EGF Mouse Model of Liver Cancer. PLoS ONE 2011, 6, e17738. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, J.; Bhar, A.; Stegmaier, P.; Kel, A.; Wingender, E. “Upstream Analysis”: An Integrated Promoter-Pathway Analysis Approach to Causal Interpretation of Microarray Data. Microarrays 2015, 4, 270–286. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.; Stegmaier, P.; Valeev, T.; Koschmann, J.; Poroikov, V.; Kel-Margoulis, O.V.; Wingender, E. Multi-omics “upstream analysis” of regulatory genomic regions helps identifying targets against methotrexate resistance of colon cancer. EuPA Open Proteom. 2016, 13, 1–13. [Google Scholar] [CrossRef]
- Volkova, Y.L.; Pickel, C.; Jucht, A.E.; Wenger, R.H.; Scholz, C.C. The Asparagine Hydroxylase FIH: A Unique Oxygen Sensor. Antioxid. Redox Signal. 2022, 37, 913–935. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidis, S.; Henze, A.T.; Kruszynska-Ziaja, I.; Skobridis, K.; Theodorou, V.; Paleolog, E.M.; Mazzone, M. Factor-inhibiting HIF-1 (FIH-1) is required for human vascular endothelial cell survival. FASEB J. 2015, 29, 2814–2827. [Google Scholar] [CrossRef]
- Tregub, P.P.; Kulikov, V.P.; Malinovskaya, N.A.; Kuzovkov, D.A.; Kovzelev, P.D. HIF-1—Alternative signals pathways of activation and formation of tolerance to hypoxia/ischemia. Patol. Fiziol. Eksp. Ter. 2019, 63, 115–122. (In Russian) [Google Scholar] [CrossRef]
- Xue, Y.; Bi, F.; Zhang, X.; Zhang, S.; Pan, Y.; Liu, N.; Shi, Y.; Yao, X.; Zheng, Y.; Fan, D. Role of Rac1 and Cdc42 in hypoxia induced p53 and von Hippel-Lindau suppression and HIF1alpha activation. Int. J. Cancer 2006, 118, 2965–2972. [Google Scholar] [CrossRef] [PubMed]
- Burbelo, P.D.; Snow, D.M.; Bahou, W.; Spiegel, S. MSE55, a Cdc42 effector protein, induces long cellular extensions in fibroblasts. Proc. Natl. Acad. Sci. USA 1999, 96, 9083–9088. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, A.J.; Calvo, F. The Borg family of Cdc42 effector proteins Cdc42EP1-5. Biochem. Soc. Trans. 2016, 44, 1709–1716. [Google Scholar] [CrossRef]
- Waleev, T.; Shtokalo, D.; Konovalova, T.; Voss, N.; Cheremushkin, E.; Stegmaier, P.; Kel-Margoulis, O.; Wingender, E.; Kel, A. Composite Module Analyst: Identification of transcription factor binding site combinations using genetic algorithm. Nucleic Acids Res. 2006, 34, W541–W545. [Google Scholar] [CrossRef] [PubMed]
- Krull, M.; Pistor, S.; Voss, N.; Kel, A.; Reuter, I.; Kronenberg, D.; Michael, H.; Schwarzer, K.; Potapov, A.; Choi, C.; et al. TRANSPATH: An information resource for storing and visualizing signaling pathways and their pathological aberrations. Nucleic Acids Res. 2006, 34, D546–D551. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Wu, J.; Li, Y.; Nie, Y.; Chen, H. Activation of MAPK participates in low shear stress-induced IL-8 gene expression in endothelial cells. Clin. Biomech. 2008, 23 (Suppl. 1), S96–S103. [Google Scholar] [CrossRef]
- Tzima, E.; del Pozo, M.A.; Shattil, S.J.; Chien, S.; Schwartz, M.A. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001, 20, 4639–4647. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Yasu, T.; Predescu, D.N.; Schmid-Schönbein, G.W. Mechanisms for Regulation of Fluid Shear Stress Response in Circulating Leukocytes. Circ. Res. 2000, 86, e13–e18. [Google Scholar] [CrossRef]
- Tinajero, M.G.; Gotlieb, A.I. Recent Developments in Vascular Adventitial Pathobiology—The Dynamic Adventitia as a Complex Regulator of Vascular Disease. Am. J. Pathol. 2020, 190, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.E.; Gössling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef]
- Boyarskikh, U.; Pintus, S.; Mandrik, N.; Stelmashenko, D.; Kiselev, I.; Evshin, I.; Sharipov, R.; Stegmaier, P.; Kolpakov, F.; Filipenko, M.; et al. Computational master-regulator search reveals mTOR and PI3K pathways responsible for low sensitivity of NCI-H292 and A427 lung cancer cell lines to cytotoxic action of p53 activator Nutlin-3. BMC Med. Genom. 2018, 11, 12. [Google Scholar] [CrossRef]
- Michael, H.; Hogan, J.; Kel, A.; Kel-Margoulis, O.; Schacherer, F.; Voss, N.; Wingender, E. Building a knowledge base for systems pathology. Brief Bioinform. 2008, 9, 518–531. [Google Scholar] [CrossRef]
- Relja, B.; Schwestka, B.; Lee, V.S.; Henrich, D.; Czerny, C.; Borsello, T.; Marzi, I.; Lehnert, M. Inhibition of c-Jun N-terminal kinase after hemorrhage but before resuscitation mitigates hepatic damage and inflammatory response in male rats. Shock 2009, 32, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, J.Y.; Dorman, B.; Zeineddin, A.; Kozar, R.A. c-Jun-mediated miR-19b expression induces endothelial barrier dysfunction in an in vitro model of hemorrhagic shock. Mol. Med. 2022, 28, 123. [Google Scholar] [CrossRef]
- Coskun, M.; Troelsen, J.T.; Nielsen, O.H. The role of CDX2 in intestinal homeostasis and inflammation. Biochim. Biophys. Acta 2011, 1812, 283–289. [Google Scholar] [CrossRef]
- Dunk, C.; Petkovic, L.; Baczyk, D.; Rossant, J.; Winterhager, E.; Lye, S. A novel in vitro model of trophoblast-mediated decidual blood vessel remodeling. Lab. Investig. 2003, 83, 1821–1828. [Google Scholar] [CrossRef]
- Zhao, H.; Jin, S.; Fan, F.; Fan, W.; Tong, T.; Zhan, Q. Activation of the transcription factor Oct-1 in response to DNA damage. Cancer Res. 2000, 60, 6276–6280. [Google Scholar]
- Schwachtgen, J.L.; Remacle, J.E.; Janel, N.; Brys, R.; Huylebroeck, D.; Meyer, D.; Kerbiriou-Nabias, D. Oct-1 is involved in the transcriptional repression of the von willebrand factor gene promoter. Blood 1998, 92, 1247–1258. [Google Scholar] [CrossRef]
- Jeong, G.A.; Choi, E.T.; Chang, J.H. Octamer-binding transcription factor-1 gene is upregulated in primary varicose veins. Ann. Vasc. Surg. 2008, 22, 115–120. [Google Scholar] [CrossRef]
- Thum, T.; Haverich, A.; Borlak, J. Cellular dedifferentiation of endothelium is linked to activation and silencing of certain nuclear transcription factors: Implications for endothelial dysfunction and vascular biology. FASEB J. 2000, 14, 740–751. [Google Scholar] [CrossRef]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zeng, J.; Wang, J.; Cui, Y.; Song, X.; Zhang, Y.; Cheng, X.; Hou, N.; Teng, Y.; Lan, Y.; et al. Hepatocyte growth factor-regulated tyrosine kinase substrate is essential for endothelial cell polarity and cerebrovascular stability. Cardiovasc. Res. 2021, 117, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Ferns, G.A.; Sprugel, K.H.; Seifert, R.A.; Bowen-Pope, D.F.; Kelly, J.D.; Murray, M.; Raines, E.W.; Ross, R. Relative platelet-derived growth factor receptor subunit expression determines cell migration to different dimeric forms of PDGF. Growth Factors 1990, 3, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Kenagy, R.D.; Fukai, N.; Min, S.K.; Jalikis, F.; Kohler, T.R.; Clowes, A.W. Proliferative capacity of vein graft smooth muscle cells and fibroblasts in vitro correlates with graft stenosis. J. Vasc. Surg. 2009, 49, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Rodt, S.A.; Ahlén, K.; Berg, A.; Rubin, K.; Reed, R.K. A novel physiological function for platelet-derived growth factor-BB in rat dermis. J. Physiol. 1996, 495 Pt 1, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Hadoke, P.W.; Wu, J.; Vesey, A.T.; Lerman, D.A.; Dweck, M.R.; Newby, D.E.; Smith, L.B.; MacRae, V.E. Ablation of the androgen receptor from vascular smooth muscle cells demonstrates a role for testosterone in vascular calcification. Sci. Rep. 2016, 6, 24807. [Google Scholar] [CrossRef] [PubMed]
- Chignalia, A.Z.; Schuldt, E.Z.; Camargo, L.L.; Montezano, A.C.; Callera, G.E.; Laurindo, F.R.; Lopes, L.R.; Avellar, M.C.; Carvalho, M.H.; Fortes, Z.B.; et al. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src-dependent pathways. Hypertension 2012, 59, 1263–1271. [Google Scholar] [CrossRef]
- Akishita, M.; Hashimoto, M.; Ohike, Y.; Ogawa, S.; Iijima, K.; Eto, M.; Ouchi, Y. Low testosterone level is an independent determinant of endothelial dysfunction in men. Hypertens. Res. 2007, 30, 1029–1034. [Google Scholar] [CrossRef]
- Rech, C.M.; Clapauch, R.; de Souza, M.; Bouskela, E. Low testosterone levels are associated with endothelial dysfunction in oophorectomized early postmenopausal women. Eur. J. Endocrinol. 2016, 174, 297–306. [Google Scholar] [CrossRef]
- Lopes, R.A.; Neves, K.B.; Pestana, C.R.; Queiroz, A.L.; Zanotto, C.Z.; Chignalia, A.Z.; Valim, Y.M.; Silveira, L.R.; Curti, C.; Tostes, R.C. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1485–H1494. [Google Scholar] [CrossRef]
- García-Honduvilla, N.; Asúnsolo, Á.; Ortega, M.A.; Sainz, F.; Leal, J.; Lopez-Hervas, P.; Pascual, G.; Buján, J. Increase and Redistribution of Sex Hormone Receptors in Premenopausal Women Are Associated with Varicose Vein Remodelling. Oxidative Med. Cell. Longev. 2018, 2018, 3974026. [Google Scholar] [CrossRef]
- Zeng, L.; Liao, H.; Liu, Y.; Lee, T.S.; Zhu, M.; Wang, X.; Stemerman, M.B.; Zhu, Y.; Shyy, J.Y. Sterol-responsive element-binding protein (SREBP) 2 down-regulates ATP-binding cassette transporter A1 in vascular endothelial cells: A novel role of SREBP in regulating cholesterol metabolism. J. Biol. Chem. 2004, 279, 48801–48807. [Google Scholar] [CrossRef]
- Xiao, H.; Lu, M.; Lin, T.Y.; Chen, Z.; Chen, G.; Wang, W.C.; Marin, T.; Shentu, T.P.; Wen, L.; Gongol, B.; et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 2013, 128, 632–642. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, H.; Su, X.; Cao, A.; Chen, F.; Chen, P.; Yan, F.; Hu, H. SREBP2 promotes the viability, proliferation, and migration and inhibits apoptosis in TGF-β1-induced airway smooth muscle cells by regulating TLR2/NF-κB/NFATc1/ABCA1 regulatory network. Bioengineered 2022, 13, 3137–3147. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, J.; Shi, B.; Wang, X.; Lu, Q.; Li, C.; Chen, H. The transcription factor LEF1 promotes tumorigenicity and activates the TGF-β signaling pathway in esophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 304. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhang, C.; Li, X.; Jia, C.; Chen, L.; Yuan, Y.; Gao, Q.; Lu, Z.; Feng, Y.; Zhao, R.; et al. LEF1 Enhances the Progression of Colonic Adenocarcinoma via Remodeling the Cell Motility Associated Structures. Int. J. Mol. Sci. 2021, 22, 10870. [Google Scholar] [CrossRef]
- Nguyen, H.; Hiscott, J.; Pitha, P.M. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997, 8, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, H.; Chao, Y.; Gu, Y.; Zhang, J.; Wang, F.; Yu, W.; Ye, P.; Chu, P.; Kong, X.; et al. Akt phosphorylation regulated by IKKε in response to low shear stress leads to endothelial inflammation via activating IRF3. Cell Signal. 2021, 80, 109900. [Google Scholar] [CrossRef] [PubMed]
- Koutsouki, E.; Beeching, C.A.; Slater, S.C.; Blaschuk, O.W.; Sala-Newby, G.B.; George, S.J. N-cadherin-dependent cell-cell contacts promote human saphenous vein smooth muscle cell survival. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Lyon, C.A.; Koutsouki, E.; Aguilera, C.M.; Blaschuk, O.W.; George, S.J. Inhibition of N-cadherin retards smooth muscle cell migration and intimal thickening via induction of apoptosis. J. Vasc. Surg. 2010, 52, 1301–1309. [Google Scholar] [CrossRef]
- Biyashev, D.; Veliceasa, D.; Topczewski, J.; Topczewska, J.M.; Mizgirev, I.; Vinokour, E.; Reddi, A.L.; Licht, J.D.; Revskoy, S.Y.; Volpert, O.V. miR-27b controls venous specification and tip cell fate. Blood 2012, 119, 2679–2687. [Google Scholar] [CrossRef]
- Glienke, J.; Fenten, G.; Seemann, M.; Sturz, A.; Thierauch, K.H. Human SPRY2 inhibits FGF2 signalling by a secreted factor. Mech. Dev. 2000, 96, 91–99. [Google Scholar] [CrossRef]
- Deng, H.; Min, E.; Baeyens, N.; Coon, B.G.; Hu, R.; Zhuang, Z.W.; Chen, M.; Huang, B.; Afolabi, T.; Zarkada, G.; et al. Activation of Smad2/3 signaling by low fluid shear stress mediates artery inward remodeling. Proc. Natl. Acad. Sci. USA 2021, 118, e2105339118. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Pirotton, S.; Communi, D.; Motte, S.; Janssens, R.; Boeynaems, J.M. Endothelial P2-purinoceptors: Subtypes and signal transduction. J. Auton. Pharmacol. 1996, 16, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; DiCorleto, P.E. ADP stimulates human endothelial cell migration via P2Y1 nucleotide receptor-mediated mitogen-activated protein kinase pathways. Circ. Res. 2008, 102, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Mato-Basalo, R.; Morente-López, M.; Arntz, O.J.; van de Loo, F.A.J.; Fafián-Labora, J.; Arufe, M.C. Therapeutic Potential for Regulation of the Nuclear Factor Kappa-B Transcription Factor p65 to Prevent Cellular Senescence and Activation of Pro-Inflammatory in Mesenchymal Stem Cells. Int. J. Mol. Sci. 2021, 22, 3367. [Google Scholar] [CrossRef]
- Bracamonte, M.P.; Jayachandran, M.; Rud, K.S.; Miller, V.M. Acute effects of 17beta -estradiol on femoral veins from adult gonadally intact and ovariectomized female pigs. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2389–H2396. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Timmons, P.M.; Hébert, J.M.; Rigby, P.W.; Tjian, R. Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev. 1991, 5, 105–119. [Google Scholar] [CrossRef]
- Tam, J.C.; Ko, C.H.; Koon, C.M.; Cheng, Z.; Lok, W.H.; Lau, C.P.; Leung, P.C.; Fung, K.P.; Chan, W.Y.; Lau, C.B. Identification of Target Genes Involved in Wound Healing Angiogenesis of Endothelial Cells with the Treatment of a Chinese 2-Herb Formula. PLoS ONE 2015, 10, e0139342. [Google Scholar] [CrossRef]
- Baryła, I.; Kośla, K.; Bednarek, A.K. WWOX and metabolic regulation in normal and pathological conditions. J. Mol. Med. 2022, 100, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, M.; Baldassari, S.; Tochigi, Y.; Kośla, K.; Buffelli, F.; Torella, A.; Severino, M.; Paladini, D.; Mandarà, L.; Riva, A.; et al. Loss of Wwox Perturbs Neuronal Migration and Impairs Early Cortical Development. Front. Neurosci. 2020, 14, 644. [Google Scholar] [CrossRef]
- Pezeshkpoor, B.; Pavlova, A.; Oldenburg, J.; El-Maarri, O. F8 genetic analysis strategies when standard approaches fail. Hamostaseologie 2014, 34, 167–173. [Google Scholar] [CrossRef]
- Wang, K.C.; Brooks, D.A.; Thornburg, K.L.; Morrison, J.L. Activation of IGF-2R stimulates cardiomyocyte hypertrophy in the late gestation sheep fetus. J. Physiol. 2012, 590, 5425–5437. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.Y.; Liu, F.; Fang, B.B.; Tian, T.; Li, Y.H.; Zhang, T.; Li, X.M.; Yang, Y.N. NFKB1 Gene Mutant Was Associated with Prognosis of Coronary Artery Disease and Exacerbated Endothelial Mitochondrial Fission and Dysfunction. Oxidative Med. Cell. Longev. 2022, 2022, 9494926. [Google Scholar] [CrossRef]
- Batchu, S.N.; Xia, J.; Ko, K.A.; Doyley, M.M.; Abe, J.; Morrell, C.N.; Korshunov, V.A. Axl modulates immune activation of smooth muscle cells in vein graft remodeling. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1048–H1058. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Gordon, D.M.; Pain, J.; Stemmler, T.L.; Dancis, A.; Pain, D. Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J. Biol. Chem. 2013, 288, 36773–36786. [Google Scholar] [CrossRef]
- Koutnikova, H.; Campuzano, V.; Foury, F.; Dollé, P.; Cazzalini, O.; Koenig, M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 1997, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Wang, J.; Zhang, R.; Jiang, F.; Tang, S.; Chen, M.; Yan, J.; Sun, X.; Wang, S.; Wang, T.; et al. Common variants in or near ZNRF1, COLEC12, SCYL1BP1 and API5 are associated with diabetic retinopathy in Chinese patients with type 2 diabetes. Diabetologia 2015, 58, 1231–1238. [Google Scholar] [CrossRef]
- Araki, T.; Nagarajan, R.; Milbrandt, J. Identification of genes induced in peripheral nerve after injury. Expression profiling and novel gene discovery. J. Biol. Chem. 2001, 276, 34131–34141. [Google Scholar] [CrossRef] [PubMed]
- Zagabathina, S.; Ramadoss, R.; Ah, H.P.; Krishnan, R. Comparative Evaluation of SMAD-2 Expression in Oral Submucous Fibrosis and Reactive Oral Lesions. Asian Pac. J. Cancer Prev. 2020, 21, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Federspiel, J.; Kollmann, C.; Teping, P.; Schwab, T.; Schäfers, H.J. SMAD3 contributes to ascending aortic dilatation independent of transforming growth factor-beta in bicuspid and unicuspid aortic valve disease. Sci. Rep. 2022, 12, 15476. [Google Scholar] [CrossRef]
- Uddin, M.; Ratanatharathorn, A.; Armstrong, D.; Kuan, P.F.; Aiello, A.E.; Bromet, E.J.; Galea, S.; Koenen, K.C.; Luft, B.; Ressler, K.J.; et al. Epigenetic meta-analysis across three civilian cohorts identifies NRG1 and HGS as blood-based biomarkers for post-traumatic stress disorder. Epigenomics 2018, 10, 1585–1601. [Google Scholar] [CrossRef]
- Lurie, F.; Passman, M.; Meisner, M.; Dalsing, M.; Masuda, E.; Welch, H.; Bush, R.L.; Blebea, J.; Carpentier, P.H.; De Maeseneer, M.; et al. The 2020 update of the CEAP classification system and reporting standards. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Genome Studio Methylation Module v1.8 User Guide (11319130). Available online: https://support.illumina.com/content/dam/illuminasupport/documents/documentation/software_documentation/genomestudio/genomestudio-2011-1/genomestudio-methylation-v1-8-user-guide-11319130-b.pdf (accessed on 22 December 2022).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Description | DiffScore 1 | ECs + Avg_Beta 2 | ECs − Avg_Beta | TargetID 3 | p-Value 4 |
|---|---|---|---|---|---|---|
| EEF1D | eukaryotic translation elongation factor 1 delta | −109.1854 | 0.6555644 | 0.8999788 | cg22186533 | 1.21 × 10−11 |
| MMD | monocyte to macrophage differentiation-associated | −35.18965 | 0.2981108 | 0.5100111 | cg14861570 | 0.000302716 |
| TESK1 | testis associated actin remodeling kinase 1 | −31.42026 | 0.4998818 | 0.6956196 | cg09000510 | 0.000721064 |
| SLC24A4 | solute carrier family 24 member 4 | −28.64084 | 0.3270859 | 0.5232344 | cg15052901 | 0.001367464 |
| MYH3 | myosin heavy chain 3 | −28.24431 | 0.2393593 | 0.4242639 | cg18190433 | 0.001498197 |
| PDGFB | platelet derived growth factor subunit B | −24.95273 | 0.11867 | 0.2644652 | cg19167673 | 0.003196885 |
| AR | androgen receptor | −24.15009 | 0.5732113 | 0.7435369 | cg27271368 | 0.003845838 |
| ZCCHC10 | zinc finger CCHC-type containing 10 | −22.23847 | 0.585928 | 0.748945 | cg08801754 | 0.005972457 |
| CELSR3 | cadherin EGF LAG seven-pass G-type receptor 3 | −22.23847 | 0.5043565 | 0.6789438 | cg06621358 | 0.005972457 |
| CNIH2 | cornichon family AMPA receptor auxiliary protein 2 | −21.25318 | 0.1661903 | 0.3161516 | cg19026260 | 0.007493453 |
| ZFP28 | ZFP28 zinc finger protein | 16.8081 | 0.4203998 | 0.2640518 | cg23850212 | 0.02085403 |
| YY1 | YY1 transcription factor | 18.57949 | 0.7956405 | 0.6552107 | cg22763181 | 0.013869187 |
| FRMD1 | FERM domain containing 1 | 19.00112 | 0.6587915 | 0.49145 | cg00350478 | 0.012586008 |
| MEST | mesoderm specific transcript | 21.27902 | 0.8093046 | 0.6647319 | cg13917504 | 0.007449 |
| LARP1 | La ribonucleoprotein 1, translational regulator | 23.04286 | 0.5051906 | 0.3232486 | cg23613317 | 0.004962654 |
| CRELD2 | cysteine rich with EGF like domains 2 | 23.1515 | 0.836981 | 0.6953279 | cg25882056 | 0.004840052 |
| RAB9A | RAB9A, member RAS oncogene family | 25.78106 | 0.7809207 | 0.6158033 | cg02620228 | 0.002641764 |
| KRT17 | keratin 17 | 28.64084 | 0.7502007 | 0.5708218 | cg00214794 | 0.001367464 |
| SLC29A1 | solute carrier family 29 member 1 (Augustine blood group) | 45.2453 | 0.7506605 | 0.5342799 | cg10519140 | 2.98862 × 10−5 |
| SLC45A1 | solute carrier family 45 member 1 | 57.2597 | 0.6744524 | 0.4233551 | cg11283860 | 1.87945 × 10−6 |
| Gene Symbol | Gene Description | DiffScore 1 | SMCs + Avg_Beta 2 | SMCs − Avg_Beta | TargetID 3 | p-Value 4 |
|---|---|---|---|---|---|---|
| SCT | secretin | −44.15369 | 0.3001944 | 0.5255814 | cg05782292 | 3.84265 × 10−5 |
| HLTF | helicase-like transcription factor | −37.04874 | 0.3306479 | 0.5443499 | cg26151310 | 0.0001973 |
| CDH2 | cadherin 2 | −34.43425 | 0.3850524 | 0.5944616 | cg09313439 | 0.000360226 |
| ZNRF1 | zinc and ring finger 1 | −30.55943 | 0.385322 | 0.5856031 | cg20957193 | 0.000879138 |
| PRADC1 | protease associated domain containing 1 | −28.45422 | 0.4262078 | 0.6192294 | cg26993951 | 0.001427506 |
| CCNJL | cyclin J-like | −21.76985 | 0.4390145 | 0.6126426 | cg17178888 | 0.006652961 |
| METTL25B | methyltransferase-like 25B | −21.18877 | 0.4171189 | 0.590144 | cg23881601 | 0.007605416 |
| CDCA5 | cell division cycle-associated 5 | −21.18382 | 0.4315823 | 0.6038101 | cg20252016 | 0.00761409 |
| SIRT3 | sirtuin 3 | −20.95826 | 0.7362766 | 0.8588417 | cg23530288 | 0.008019993 |
| TCF15 | transcription factor 15 | −20.04535 | 0.3651721 | 0.534321 | cg06143901 | 0.009896121 |
| TMEM215 | transmembrane protein 215 | 29.97724 | 0.8431917 | 0.6911527 | cg11308840 | 0.001005254 |
| KMT2D | lysine methyltransferase 2D | 30.48615 | 0.4764972 | 0.2821061 | cg24471867 | 0.000894098 |
| CDC37L1 | cell division cycle 37-like 1, HSP90 cochaperone | 30.55943 | 0.8350812 | 0.6772436 | cg22189519 | 0.000879138 |
| SPA17 | sperm autoantigenic protein 17 | 34.0979 | 0.8123621 | 0.6387361 | cg22318304 | 0.000389233 |
| PGBD4 | piggyBac transposable element derived 4 | 35.79305 | 0.6302417 | 0.4193449 | cg12237946 | 0.000263448 |
| KHK | ketohexokinase | 39.8873 | 0.8598925 | 0.6937668 | cg01522194 | 0.000102629 |
| RNF213 | ring finger protein 213 | 44.15369 | 0.8767144 | 0.7105584 | cg09907395 | 3.84265 × 10−5 |
| MRPL12 | mitochondrial ribosomal protein L12 | 48.63778 | 0.816642 | 0.6133671 | cg01372689 | 1.36843 × 10−5 |
| RPRM | reprimo, TP53-dependent G2 arrest mediator homolog | 58.14957 | 0.6450388 | 0.3928624 | cg18411898 | 1.53124 × 10−6E |
| SLC22A18 | solute carrier family 22 member 18 | 77.42734 | 0.8139806 | 0.5674041 | cg16035277 | 1.80828 × 10−8 |
| Gene Symbol | Gene Description | DiffScore 1 | FBs + Avg_Beta 2 | FBs − Avg_Beta | TargetID 3 | p-Value 4 |
|---|---|---|---|---|---|---|
| PPP1R13L | protein phosphatase 1 regulatory subunit 13-like | −287.4783 | 0.1425181 | 0.5611927 | cg03554552 | 1.78719 × 10−29 |
| PPP1CC | protein phosphatase 1 catalytic subunit gamma | −195.8575 | 0.5820595 | 0.9008529 | cg03310453 | 2.59567 × 10−20 |
| SCRN2 | secernin 2 | −174.5741 | 0.5376809 | 0.858446 | cg11646887 | 3.48811 × 10−18 |
| MYL5 | myosin light chain 5 | −174.3685 | 0.5000765 | 0.831169 | cg18176712 | 3.65721 × 10−18 |
| FUCA1 | alpha-L-fucosidase 1 | −166.3722 | 0.6312342 | 0.9132504 | cg24792360 | 2.30558 × 10−17 |
| ISYNA1 | inositol-3-phosphate synthase 1 | −161.8102 | 0.4622425 | 0.7933455 | cg09374949 | 6.59144 × 10−17 |
| YIPF5 | Yip1 domain family member 5 | −153.572 | 0.4413335 | 0.7704819 | cg04293726 | 4.39339 × 10−16 |
| SEMA6C | semaphorin 6C | −149.7812 | 0.4570227 | 0.7800868 | cg25820693 | 1.05167 × 10−15 |
| FBN2 | fibrillin 2 | −147.2405 | 0.7476242 | 0.9649287 | cg25084878 | 1.88777 × 10−15 |
| RANGNRF | RAN guanine nucleotide release factor | −146.2499 | 0.4167334 | 0.743665 | cg11178443 | 2.37143 × 10−15 |
| DCAF16 | DDB1 and CUL4-associated factor 16 | 90.26163 | 0.7206882 | 0.4538092 | cg27127056 | 9.41536 × 10−10 |
| SACM1L | SAC1-like phosphatidylinositide phosphatase | 91.75178 | 0.8458125 | 0.6134535 | cg22986271 | 6.6807 × 10−10 |
| CACHD1 | cache domain containing 1 | 94.27448 | 0.7917484 | 0.5357191 | cg19304410 | 3.73725 × 10−10 |
| UGDH | UDP-glucose 6-dehydrogenase | 97.15292 | 0.8846164 | 0.6643396 | cg25117976 | 1.92623 × 10−10 |
| GGTLC2 | gamma-glutamyltransferase light chain 2 | 99.88158 | 0.8204779 | 0.5666317 | cg01457622 | 1.02764 × 10−10 |
| C16orf54 | chromosome 16 open reading frame 54 | 101.7236 | 0.2649694 | 0.0511992 | cg23093496 | 6.72419 × 10−11 |
| FXN | frataxin | 103.5087 | 0.5243153 | 0.2395225 | cg23667933 | 4.4579 × 10−11 |
| RGS11 | regulator of G protein signaling 11 | 104.2319 | 0.4908251 | 0.2102013 | cg01344518 | 3.77407 × 10−11 |
| PRICKLE3 | prickle planar cell polarity protein 3 | 105.7841 | 0.3453371 | 0.09902742 | cg10417559 | 2.63992 × 10−11 |
| SPATA4 | spermatogenesis associated 4 | 335.9589 | 0.5617582 | 0.2024512 | cg01311285 | 2.53577 × 10−34 |
| Gene Symbol | Gene Description | Regulatory Score 1 | Yes-No Ratio 2 |
|---|---|---|---|
| ECs | |||
| JUN | Jun proto-oncogene, AP-1 transcription factor subunit | 2.09 | 1.56 |
| CDX2 | caudal type homeobox 2 | 1.77 | 1.62 |
| POU2F1 | POU class 2 homeobox 1 | 1.61 | 1.87 |
| SREBF2 | sterol regulatory element binding transcription factor 2 | 1.59 | 1.33 |
| NKX3-1 | NK3 homeobox 1 | 1.58 | 1.58 |
| NHLH1 | nescient helix-loop-helix 1 | 1.39 | 2.6 |
| GABPA | GA binding protein transcription factor subunit alpha | 1.38 | 1.24 |
| RUNX3 | RUNX family transcription factor 3 | 1.32 | 4.61 |
| NRL | neural retina leucine zipper | 1.21 | 1.75 |
| MAFK | MAF bZIP transcription factor K | 1.15 | 2.13 |
| SMCs | |||
| SREBF2 | sterol regulatory element binding transcription factor 2 | 1.8 | 1.5 |
| LEF1 | lymphoid enhancer binding factor 1 | 1.78 | 2.26 |
| IRF3 | interferon regulatory factor 3 | 1.76 | 2.39 |
| WT1 | WT1 transcription factor | 1.74 | 5.13 |
| SFPQ | splicing factor proline and glutamine rich | 1.71 | 1.24 |
| POU2F1 | POU class 2 homeobox 1 | 1.7 | 3.54 |
| RBPJ | recombination signal binding protein for immunoglobulin kappa J region | 1.7 | 1.65 |
| DPF2 | double PHD fingers 2 | 1.64 | 1.31 |
| PAX5 | paired box 5 | 1.64 | 1.35 |
| SREBF1 | sterol regulatory element binding transcription factor 1 | 1.64 | 4.45 |
| FBs | |||
| RELA | RELA proto-oncogene, NF-kB subunit | 2.27 | 1.9 |
| ESR1 | estrogen receptor 1 | 2.09 | 2.44 |
| TFAP2A | transcription factor AP-2 alpha | 1.66 | 2.12 |
| RUNX3 | RUNX family transcription factor 3 | 1.62 | 1.8 |
| ELF3 | E74 like ETS transcription factor 3 | 1.46 | 2.96 |
| TFAP2B | transcription factor AP-2 beta | 0 | 2.12 |
| TFAP2C | transcription factor AP-2 gamma | 0 | 2.42 |
| Master Molecule Name | Gene Symbol | Gene Description | Total Rank 1 |
|---|---|---|---|
| ECs | |||
| PDGFB-isoform1(h) | PDGFB | platelet-derived growth factor subunit B | 9 |
| PDGFB-isoform2(h) | PDGFB | platelet-derived growth factor subunit B | 9 |
| PDGFB(h) | PDGFB | platelet-derived growth factor subunit B | 11 |
| HRS(h) | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 13 |
| HRS:PtdIns(3)P:SARA:SMAD2 | HGS, SMAD2, ZFYVE9 | SMAD family member 2, hepatocyte growth factor-regulated tyrosine kinase substrate, zinc finger FYVE-type containing 9 | 13 |
| HRS:PtdIns(3)P:SARA:SMAD3 | HGS, SMAD3, ZFYVE9 | SMAD family member 3, hepatocyte growth factor-regulated tyrosine kinase substrate, zinc finger FYVE-type containing 9 | 13 |
| HRS-isoform1(h) | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 13 |
| HRS(h){pY} | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 13 |
| HRS-isoform2(h) | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 13 |
| HRS(h){pY334} | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 13 |
| SMCs | |||
| HRS(h) | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 30 |
| HRS:PtdIns(3)P:SARA:SMAD2 | HGS, SMAD2, ZFYVE9 | SMAD family member 2, hepatocyte growth factor-regulated tyrosine kinase substrate, zinc finger FYVE-type containing 9 | 30 |
| HRS:PtdIns(3)P:SARA:SMAD3 | HGS, SMAD3, ZFYVE9 | SMAD family member 3, hepatocyte growth factor-regulated tyrosine kinase substrate, zinc finger FYVE-type containing 9 | 30 |
| HRS-isoform1(h) | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 30 |
| HRS(h){pY} | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 30 |
| HRS-isoform2(h) | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 30 |
| HRS(h){pY334} | HGS | hepatocyte growth factor-regulated tyrosine kinase substrate | 30 |
| Sprouty2(h){p} | SPRY2 | sprouty RTK signaling antagonist 2 | 30 |
| Sprouty2(h) | SPRY2 | sprouty RTK signaling antagonist 2 | 31 |
| CD23(h) | FCER2 | Fc fragment of IgE receptor II | 32 |
| FBs | |||
| SOCS-1(h) | SOCS1 | suppressor of cytokine signaling 1 | 21 |
| p50:NF-kappaBp65:SOCS-1 | NFKB1, RELA, SOCS1 | RELA proto-oncogene, NF-kB subunit, nuclear factor kappa B subunit 1, suppressor of cytokine signaling 1 | 21 |
| p50:NF-kappaBp65{ub}n:SOCS-1 | NFKB1, RELA, SOCS1 | RELA proto-oncogene, NF-kB subunit, nuclear factor kappa B subunit 1, suppressor of cytokine signaling 1 | 21 |
| SOCS-1(h) | SOCS1 | suppressor of cytokine signaling 1 | 29 |
| IGF-2R(h) | IGF2R | insulin like growth factor 2 receptor | 32 |
| G12/13 | GNA12, GNA13, GNB1, GNB1L, GNB2, GNB3, GNB4, GNB5, GNG2, GNG3, GNG5, GNGT1 | G protein subunit alpha 12, G protein subunit alpha 13, G protein subunit beta 1, G protein subunit beta 1 like, G protein subunit beta 2, G protein subunit beta 3, G protein subunit beta 4, G protein subunit beta 5, G protein subunit gamma 2, G protein subunit gamma 3, G protein subunit gamma 5, G protein subunit gamma transducin 1 | 49 |
| DHTXisoform1(h) | UVRAG | UV radiation resistance associated | 50 |
| F8B(h){gly}n | F8 | coagulation factor VIII | 51 |
| F8B(h) | F8 | coagulation factor VIII | 52 |
| F8B-isoform2(h) | F8 | coagulation factor VIII | 53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smetanina, M.A.; Korolenya, V.A.; Kel, A.E.; Sevostyanova, K.S.; Gavrilov, K.A.; Shevela, A.I.; Filipenko, M.L. Epigenome-Wide Changes in the Cell Layers of the Vein Wall When Exposing the Venous Endothelium to Oscillatory Shear Stress. Epigenomes 2023, 7, 8. https://doi.org/10.3390/epigenomes7010008
Smetanina MA, Korolenya VA, Kel AE, Sevostyanova KS, Gavrilov KA, Shevela AI, Filipenko ML. Epigenome-Wide Changes in the Cell Layers of the Vein Wall When Exposing the Venous Endothelium to Oscillatory Shear Stress. Epigenomes. 2023; 7(1):8. https://doi.org/10.3390/epigenomes7010008
Chicago/Turabian StyleSmetanina, Mariya A., Valeria A. Korolenya, Alexander E. Kel, Ksenia S. Sevostyanova, Konstantin A. Gavrilov, Andrey I. Shevela, and Maxim L. Filipenko. 2023. "Epigenome-Wide Changes in the Cell Layers of the Vein Wall When Exposing the Venous Endothelium to Oscillatory Shear Stress" Epigenomes 7, no. 1: 8. https://doi.org/10.3390/epigenomes7010008
APA StyleSmetanina, M. A., Korolenya, V. A., Kel, A. E., Sevostyanova, K. S., Gavrilov, K. A., Shevela, A. I., & Filipenko, M. L. (2023). Epigenome-Wide Changes in the Cell Layers of the Vein Wall When Exposing the Venous Endothelium to Oscillatory Shear Stress. Epigenomes, 7(1), 8. https://doi.org/10.3390/epigenomes7010008