The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma



Abstract
1. Introduction
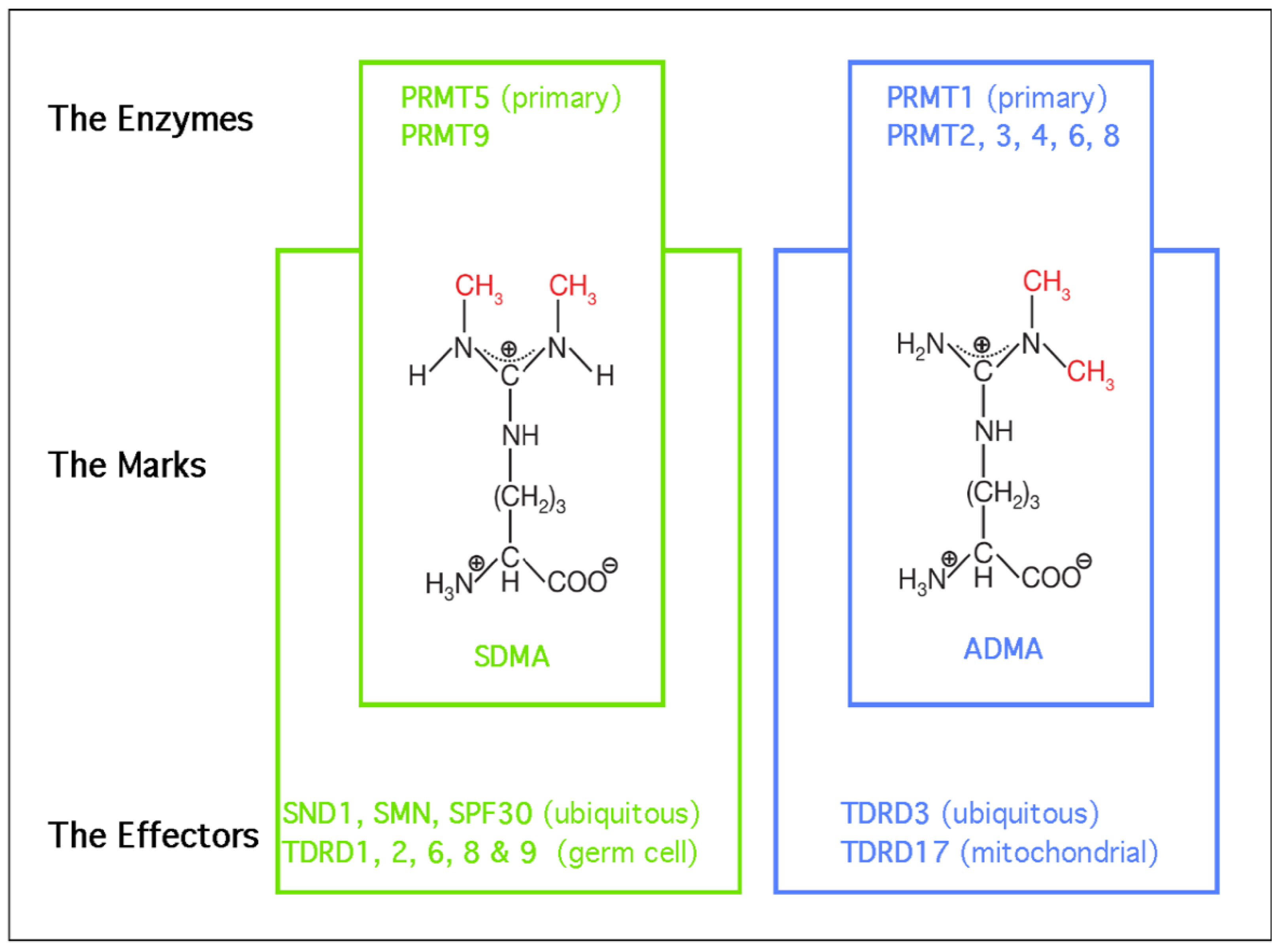
2. Biological Roles of PRMT5—The Primary Depositor of SDMA Marks
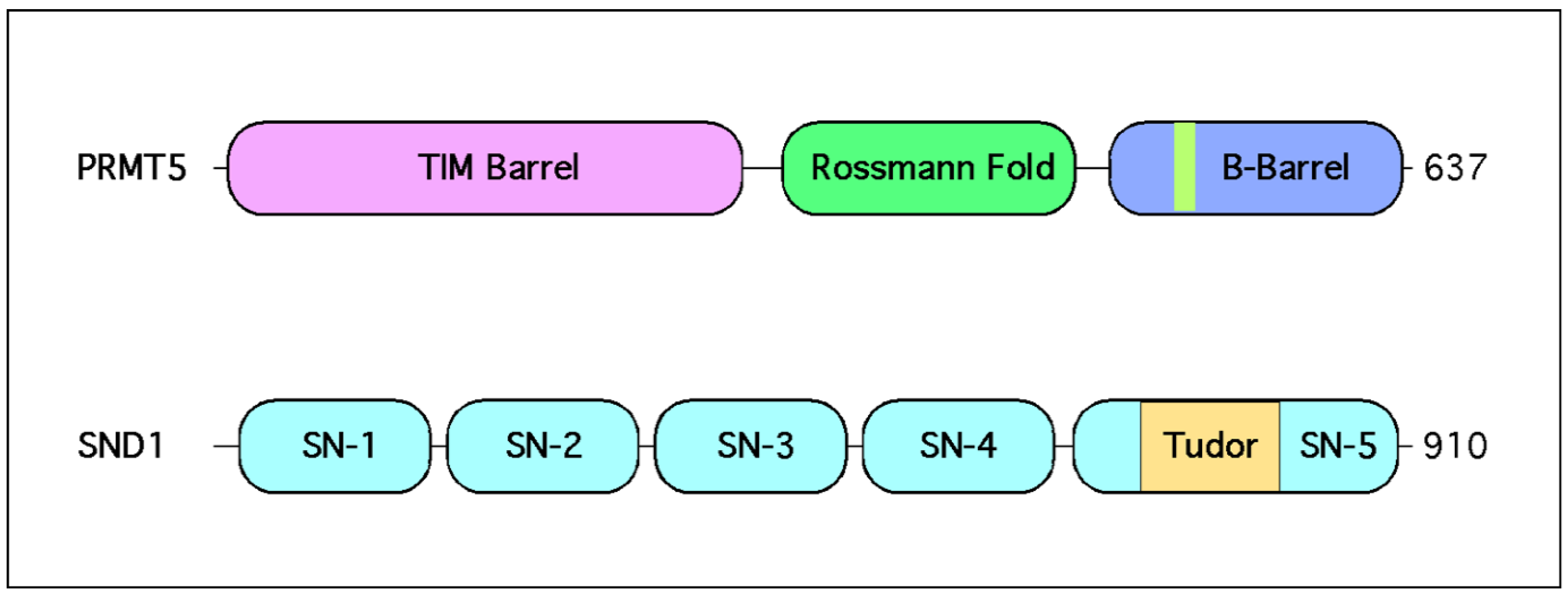
2.1. PRMT5 Forms a Stable Complex with MEP50
2.2. The Methylosome is Targeted to Distinct Substrates by Adaptor Proteins
2.3. The Identification of PRMT5 Substrates Implicate It in the Regulation of Transcription, Splicing, Signal Transduction and the Repair of DNA Damage
2.4. PRMT5 Functional Misdirection Due to Cross-Reactivity with the FLAG Antibody
2.5. Mouse Models Reveal a Number of Biological Roles for the Methylosome
3. Biological Roles of SND1—A Major Reader of SDMA Marks
3.1. The Tudor Domain of SND1 Interacts Selectively with SDMA Marks
3.2. SND1 as a Transcriptional Coactivator
3.3. SND1 Is a Splicing Factor
3.4. SND1 Regulates RNA Stability
3.5. SND1 as a Component of Exosome Cargo
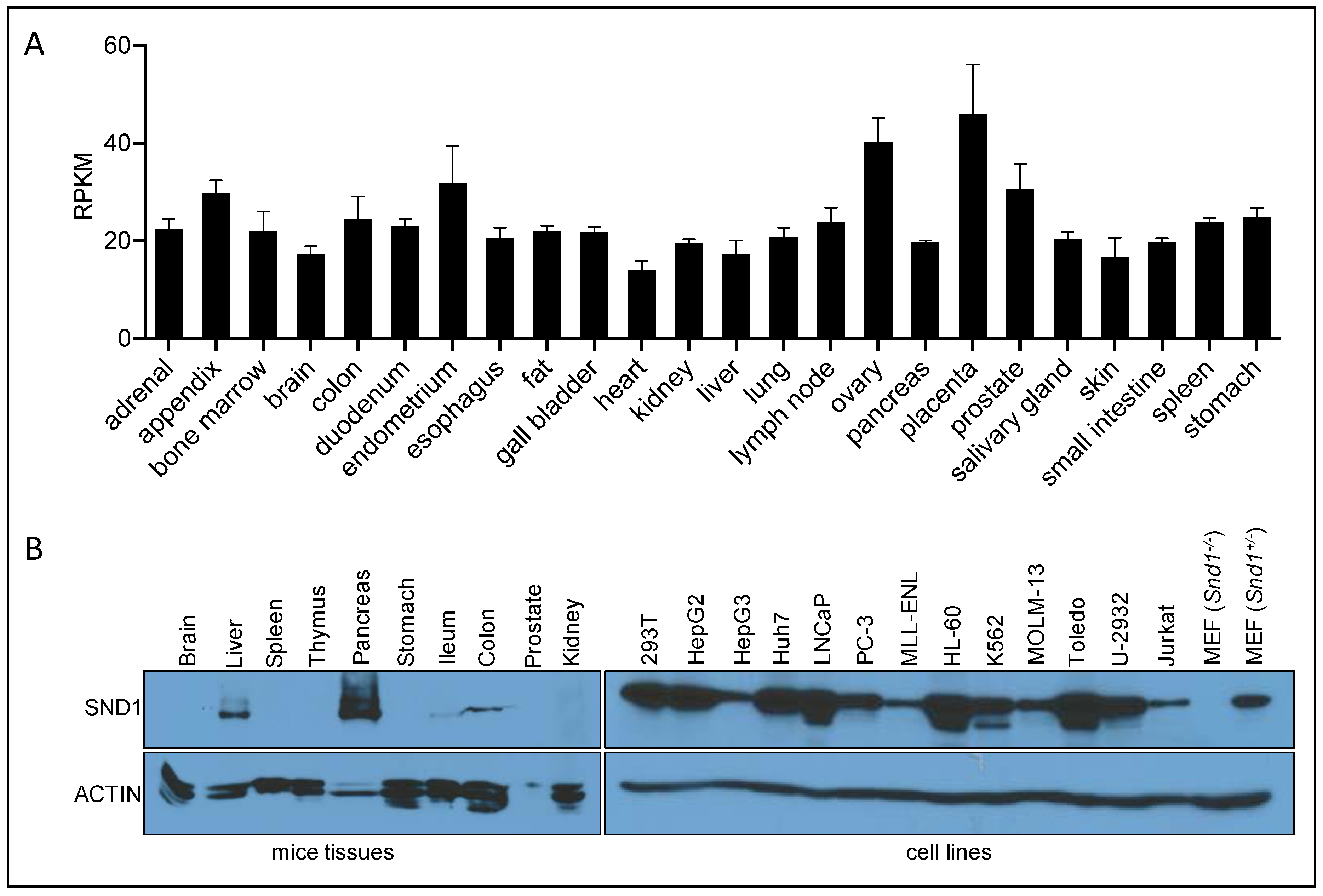
3.6. SND1 Expression Patterns at the RNA and Protein Levels
3.7. Mouse Models of SND1 Overexpression Support Its Potential Oncogenic Functions
3.8. Mouse Syngeneic Tumor Models Reveal a Role for SND1 (and PRMT5) in Antitumor Immunity
3.9. SND1 Is Likely an Oncogene
4. Hallmarks of HCC
4.1. PRMT5 and HCC
4.2. SND1 and HCC
5. Targeting Elevated SND1 Levels with PRMT5 Inhibitors
6. Conclusion and Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Chen, J.; Sagum, C.; Bedford, M.T. Protein domain microarrays as a platform to decipher signaling pathways and the histone code. Methods 2019, 184, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. NDUFAF7 methylates arginine 85 in the NDUFS2 subunit of human complex I. J. Biol. Chem. 2013, 288, 33016–33026. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hadjikyriacou, A.; Xia, Z.; Gayatri, S.; Kim, D.; Zurita-Lopez, C.; Kelly, R.; Guo, A.; Li, W.; Clarke, S.G.; et al. PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat. Commun. 2015, 6, 6428. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M. Epsilon-N-methylated lysine and guanidine-N-methylated arginine of proteins. 3. Presence and distribution in nature and mammals. Seikagaku 1972, 44, 364–370. [Google Scholar]
- Paik, W.K.; Kim, S. Natural Occurrence of Various Methylated Amino Acid Derivatives; John Wiley & Sons: New York, NY, USA, 1980; pp. 8–25. [Google Scholar]
- Dhar, S.; Vemulapalli, V.; Patananan, A.N.; Huang, G.L.; Di Lorenzo, A.; Richard, S.; Comb, M.J.; Guo, A.; Clarke, S.G.; Bedford, M.T. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep. 2013, 3, 1311. [Google Scholar] [CrossRef]
- Gayatri, S.; Bedford, M.T. Readers of histone methylarginine marks. Biochim. Biophys. Acta 2014, 1839, 702–710. [Google Scholar] [CrossRef]
- Ponting, C.P. Tudor domains in proteins that interact with RNA. Trends Biochem. Sci. 1997, 22, 51–52. [Google Scholar] [CrossRef]
- Friesen, W.J.; Massenet, S.; Paushkin, S.; Wyce, A.; Dreyfuss, G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001, 7, 1111–1117. [Google Scholar] [CrossRef]
- Selenko, P.; Sprangers, R.; Stier, G.; Buhler, D.; Fischer, U.; Sattler, M. SMN tudor domain structure and its interaction with the Sm proteins. Nat. Struct. Biol. 2001, 8, 27–31. [Google Scholar]
- Shimizu, D.; Kanda, M.; Sugimoto, H.; Shibata, M.; Tanaka, H.; Takami, H.; Iwata, N.; Hayashi, M.; Tanaka, C.; Kobayashi, D.; et al. The protein arginine methyltransferase 5 promotes malignant phenotype of hepatocellular carcinoma cells and is associated with adverse patient outcomes after curative hepatectomy. Int. J. Oncol. 2017, 50, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Jariwala, N.; Rajasekaran, D.; Mendoza, R.G.; Shen, X.N.; Siddiq, A.; Akiel, M.A.; Robertson, C.L.; Subler, M.A.; Windle, J.J.; Fisher, P.B.; et al. Oncogenic Role of SND1 in Development and Progression of Hepatocellular Carcinoma. Cancer Res. 2017, 77, 3306–3316. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, R.M.; Thompson, P.R. Substrate specificity, processivity, and kinetic mechanism of protein arginine methyltransferase 5. Biochemistry 2013, 52, 5430–5440. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Friesen, W.J.; Wyce, A.; Paushkin, S.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. A novel WD repeat protein component of the methylosome binds Sm proteins. J. Biol. Chem. 2002, 277, 8243–8247. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Dhar, S.; Bedford, M.T. PRMT5 regulates IRES-dependent translation via methylation of hnRNP A1. Nucleic Acids Res. 2017, 45, 4359–4369. [Google Scholar] [CrossRef]
- Timm, D.E.; Bowman, V.; Madsen, R.; Rauch, C. Cryo-electron microscopy structure of a human PRMT5:MEP50 complex. PLoS ONE 2018, 13, e0193205. [Google Scholar] [CrossRef]
- Ho, M.C.; Wilczek, C.; Bonanno, J.B.; Xing, L.; Seznec, J.; Matsui, T.; Carter, L.G.; Onikubo, T.; Kumar, P.R.; Chan, M.K.; et al. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS ONE 2013, 8, e57008. [Google Scholar] [CrossRef]
- Antonysamy, S.; Bonday, Z.; Campbell, R.M.; Doyle, B.; Druzina, Z.; Gheyi, T.; Han, B.; Jungheim, L.N.; Qian, Y.; Rauch, C.; et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 17960–17965. [Google Scholar] [CrossRef]
- Hosohata, K.; Li, P.; Hosohata, Y.; Qin, J.; Roeder, R.G.; Wang, Z. Purification and identification of a novel complex which is involved in androgen receptor-dependent transcription. Mol. Cell. Biol. 2003, 23, 7019–7029. [Google Scholar] [CrossRef]
- Wilczek, C.; Chitta, R.; Woo, E.; Shabanowitz, J.; Chait, B.T.; Hunt, D.F.; Shechter, D. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Biol. Chem. 2011, 286, 42221–42231. [Google Scholar] [CrossRef]
- Meister, G.; Eggert, C.; Buhler, D.; Brahms, H.; Kambach, C.; Fischer, U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001, 11, 1990–1994. [Google Scholar] [CrossRef]
- Pesiridis, G.S.; Diamond, E.; Van Duyne, G.D. Role of pICLn in methylation of Sm proteins by PRMT5. J. Biol. Chem. 2009, 284, 21347–21359. [Google Scholar] [CrossRef] [PubMed]
- Paknia, E.; Chari, A.; Stark, H.; Fischer, U. The Ribosome Cooperates with the Assembly Chaperone pICln to Initiate Formation of snRNPs. Cell Rep. 2016, 16, 3103–3112. [Google Scholar] [CrossRef] [PubMed]
- Guderian, G.; Peter, C.; Wiesner, J.; Sickmann, A.; Schulze-Osthoff, K.; Fischer, U.; Grimmler, M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 2011, 286, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Messaoudi, S.E.; Rodier, G.; Le Cam, A.; Sardet, C.; Fabbrizio, E. The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep. 2008, 9, 452–458. [Google Scholar] [CrossRef]
- Tamiya, H.; Kim, H.; Klymenko, O.; Kim, H.; Feng, Y.; Zhang, T.; Han, J.Y.; Murao, A.; Snipas, S.J.; Jilaveanu, L.; et al. SHARPIN-mediated regulation of protein arginine methyltransferase 5 controls melanoma growth. J. Clin. Investig. 2018, 128, 517–530. [Google Scholar] [CrossRef]
- Yang, M.; Lin, X.; Segers, F.; Suganthan, R.; Hildrestrand, G.A.; Rinholm, J.E.; Aas, P.A.; Sousa, M.M.L.; Holm, S.; Bolstad, N.; et al. OXR1A, a Coactivator of PRMT5 Regulating Histone Arginine Methylation. Cell Rep. 2020, 30, 4165–4178. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; McDonald, E.R., 3rd; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; deWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Pollack, B.P.; Kotenko, S.V.; He, W.; Izotova, L.S.; Barnoski, B.L.; Pestka, S. The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J. Biol. Chem. 1999, 274, 31531–31542. [Google Scholar] [CrossRef]
- Tee, W.W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; Cote, J.; Boulanger, M.C.; Richard, S. A Proteomic Analysis of Arginine-methylated Protein Complexes. Mol. Cell. Proteom. 2003, 2, 1319–1330. [Google Scholar] [CrossRef]
- Musiani, D.; Bok, J.; Massignani, E.; Wu, L.; Tabaglio, T.; Ippolito, M.R.; Cuomo, A.; Ozbek, U.; Zorgati, H.; Ghoshdastider, U.; et al. Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Nishioka, K.; Reinberg, D. Methods and tips for the purification of human histone methyltransferases. Methods 2003, 31, 49–58. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Chen, G.I.; Gingras, A.C. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods 2007, 42, 298–305. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef]
- Zhou, L.; Wu, H.; Lee, P.; Wang, Z. Roles of the androgen receptor cofactor p44 in the growth of prostate epithelial cells. J. Mol. Endocrinol. 2006, 37, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cheng, G.; Hamard, P.J.; Greenblatt, S.; Wang, L.; Man, N.; Perna, F.; Xu, H.; Tadi, M.; Luciani, L.; et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Investig. 2015, 125, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Litzler, L.C.; Zahn, A.; Meli, A.P.; Hebert, S.; Patenaude, A.M.; Methot, S.P.; Sprumont, A.; Bois, T.; Kitamura, D.; Costantino, S.; et al. PRMT5 is essential for B cell development and germinal center dynamics. Nat. Commun. 2019, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nagai, Y.; Okumura, M.; Greene, M.I.; Kambayashi, T. PRMT5 Is Required for T Cell Survival and Proliferation by Maintaining Cytokine Signaling. Front. Immunol. 2020, 11, 621. [Google Scholar] [CrossRef]
- Norrie, J.L.; Li, Q.; Co, S.; Huang, B.L.; Ding, D.; Uy, J.C.; Ji, Z.; Mackem, S.; Bedford, M.T.; Galli, A.; et al. PRMT5 is essential for the maintenance of chondrogenic progenitor cells in the limb bud. Development 2016, 143, 4608–4619. [Google Scholar] [CrossRef]
- Gao, S.; Wu, H.; Wang, F.; Wang, Z. Altered differentiation and proliferation of prostate epithelium in mice lacking the androgen receptor cofactor p44/WDR77. Endocrinology 2010, 151, 3941–3953. [Google Scholar] [CrossRef]
- Gu, Z.; Zhang, F.; Wang, Z.Q.; Ma, W.; Davis, R.E.; Wang, Z. The p44/wdr77-dependent cellular proliferation process during lung development is reactivated in lung cancer. Oncogene 2013, 32, 1888–1900. [Google Scholar] [CrossRef]
- Scaglione, A.; Patzig, J.; Liang, J.; Frawley, R.; Bok, J.; Mela, A.; Yattah, C.; Zhang, J.; Teo, S.X.; Zhou, T.; et al. PRMT5-mediated regulation of developmental myelination. Nat. Commun. 2018, 9, 2840. [Google Scholar] [CrossRef]
- Rho, J.; Choi, S.; Seong, Y.R.; Cho, W.K.; Kim, S.H.; Im, D.S. Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J. Biol. Chem. 2001, 276, 11393–11401. [Google Scholar] [CrossRef]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- Tan, D.Q.; Li, Y.; Yang, C.; Li, J.; Tan, S.H.; Chin, D.W.L.; Nakamura-Ishizu, A.; Yang, H.; Suda, T. PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep. 2019, 26, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Liu, F.; Veazey, K.J.; Gao, G.; Das, P.; Neves, L.F.; Lin, K.; Zhong, Y.; Lu, Y.; Giuliani, V.; et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018, 32, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Jensen, O.N.; Helin, K. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Fong, J.Y.; Pignata, L.; Goy, P.A.; Kawabata, K.C.; Lee, S.C.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; He, X.; Cao, Y.; O’Dwyer, K.; Szigety, K.M.; Wu, Y.; Gurung, B.; Feng, Z.; Katona, B.W.; Hua, X. Islet-specific Prmt5 excision leads to reduced insulin expression and glucose intolerance in mice. J. Endocrinol. 2020, 244, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gunther, S.; Looso, M.; Kunne, C.; Kruger, M.; Kim, J.; Zhou, Y.; Braun, T. Prmt5 is a regulator of muscle stem cell expansion in adult mice. Nat. Commun. 2015, 6, 7140. [Google Scholar] [CrossRef]
- Ramachandran, J.; Liu, Z.; Gray, R.S.; Vokes, S.A. PRMT5 is necessary to form distinct cartilage identities in the knee and long bone. Dev. Biol. 2019, 456, 154–163. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, T.; Li, Q.; Liu, C.; Han, F.; Chen, M.; Zhang, L.; Cui, X.; Qin, Y.; Bao, S.; et al. Prmt5 is required for germ cell survival during spermatogenesis in mice. Sci. Rep. 2015, 5, 11031. [Google Scholar] [CrossRef]
- Li, Q.; Jiao, J.; Li, H.; Wan, H.; Zheng, C.; Cai, J.; Bao, S. Histone arginine methylation by Prmt5 is required for lung branching morphogenesis through repression of BMP signaling. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Peng, H.; Ayyanathan, K.; Yan, K.P.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J., 3rd. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell. Biol. 2008, 28, 3198–3207. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Baiocchi, R.A.; Byrd, J.C.; Grever, M.R.; Jacob, S.T.; Sif, S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–3569. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pal, S.; Sif, S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 2008, 28, 6262–6277. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Sohn, H.Y.; Yoon, S.Y.; Oh, J.H.; Yang, J.O.; Kim, J.H.; Song, K.S.; Rho, S.M.; Yoo, H.S.; Kim, Y.S.; et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin. Cancer Res. 2005, 11, 473–482. [Google Scholar] [PubMed]
- Cho, E.C.; Zheng, S.; Munro, S.; Liu, G.; Carr, S.M.; Moehlenbrink, J.; Lu, Y.C.; Stimson, L.; Khan, O.; Konietzny, R.; et al. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012, 31, 1785–1797. [Google Scholar] [CrossRef]
- Jing, P.; Xie, N.; Zhu, X.; Dang, H.; Gu, Z. The methylation induced by protein arginine methyltransferase 5 promotes tumorigenesis and progression of lung cancer. J. Thorac. Dis. 2018, 10, 7014–7019. [Google Scholar] [CrossRef]
- Wei, T.Y.; Juan, C.C.; Hisa, J.Y.; Su, L.J.; Lee, Y.C.; Chou, H.Y.; Chen, J.M.; Wu, Y.C.; Chiu, S.C.; Hsu, C.P.; et al. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 2012, 103, 1640–1650. [Google Scholar] [CrossRef]
- Bao, X.; Zhao, S.; Liu, T.; Liu, Y.; Liu, Y.; Yang, X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J. Histochem. Cytochem. 2013, 61, 206–217. [Google Scholar] [CrossRef]
- Nicholas, C.; Yang, J.; Peters, S.B.; Bill, M.A.; Baiocchi, R.A.; Yan, F.; Sif, S.; Tae, S.; Gaudio, E.; Wu, X.; et al. PRMT5 is upregulated in malignant and metastatic melanoma and regulates expression of MITF and p27(Kip1.). PLoS ONE 2013, 8, e74710. [Google Scholar] [CrossRef]
- Han, X.; Li, R.; Zhang, W.; Yang, X.; Wheeler, C.G.; Friedman, G.K.; Province, P.; Ding, Q.; You, Z.; Fathallah-Shaykh, H.M.; et al. Expression of PRMT5 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth in vitro. J. Neurooncol. 2014, 118, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Virk, S.; Barnholtz-Sloan, J.; Bell, E.H.; Wojton, J.; et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752–1765. [Google Scholar] [CrossRef]
- Jiang, H.; Zhu, Y.; Zhou, Z.; Xu, J.; Jin, S.; Xu, K.; Zhang, H.; Sun, Q.; Wang, J.; Xu, J. PRMT5 promotes cell proliferation by inhibiting BTG2 expression via the ERK signaling pathway in hepatocellular carcinoma. Cancer Med. 2018, 7, 869–882. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Lee, J.S.; Park, E.R.; Shen, Y.N.; Kim, M.Y.; Shin, H.J.; Joo, H.Y.; Cho, E.H.; Moon, S.M.; Shin, U.S.; et al. Protein arginine methyltransferase 5 is implicated in the aggressiveness of human hepatocellular carcinoma and controls the invasive activity of cancer cells. Oncol. Rep. 2018, 40, 536–544. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, S.; Li, Z.; Lu, L.; Zhang, S.; Chen, X.; Cen, X.; Wu, Y. Targeting protein arginine methyltransferase 5 inhibits human hepatocellular carcinoma growth via the downregulation of beta-catenin. J. Transl. Med. 2015, 13, 349. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Liu, X.; Li, S.; Wang, Q.; Di, C.; Hu, Z.; Yu, T.; Ding, J.; Li, J.; et al. The LINC01138 drives malignancies via activating arginine methyltransferase 5 in hepatocellular carcinoma. Nat. Commun. 2018, 9, 1572. [Google Scholar] [CrossRef]
- Zheng, B.N.; Ding, C.H.; Chen, S.J.; Zhu, K.; Shao, J.; Feng, J.; Xu, W.P.; Cai, L.Y.; Zhu, C.P.; Duan, W.; et al. Targeting PRMT5 Activity Inhibits the Malignancy of Hepatocellular Carcinoma by Promoting the Transcription of HNF4alpha. Theranostics 2019, 9, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ki, B.S.; Hong, K.; Park, S.P.; Ko, J.J.; Choi, Y. Tudor Domain Containing Protein TDRD12 Expresses at the Acrosome of Spermatids in Mouse Testis. Asian-Australas J. Anim. Sci. 2016, 29, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Das, S.K.; Gredler, R.; Chen, D.; Srivastava, J.; Robertson, C.; Baldwin, A.S., Jr.; Fisher, P.B.; Sarkar, D. Multifunction protein staphylococcal nuclease domain containing 1 (SND1) promotes tumor angiogenesis in human hepatocellular carcinoma through novel pathway that involves nuclear factor kappaB and miR-221. J. Biol. Chem. 2012, 287, 13952–13958. [Google Scholar] [CrossRef]
- Zheng, S.; Moehlenbrink, J.; Lu, Y.C.; Zalmas, L.P.; Sagum, C.A.; Carr, S.; McGouran, J.F.; Alexander, L.; Fedorov, O.; Munro, S.; et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol. Cell 2013, 52, 37–51. [Google Scholar] [CrossRef]
- Chidambaranathan-Reghupaty, S.; Mendoza, R.; Fisher, P.B.; Sarkar, D. The multifaceted oncogene SND1 in cancer: Focus on hepatocellular carcinoma. Hepatoma Res. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Arretxe, E.; Armengol, S.; Mula, S.; Chico, Y.; Ochoa, B.; Martinez, M.J. Profiling of promoter occupancy by the SND1 transcriptional coactivator identifies downstream glycerolipid metabolic genes involved in TNFalpha response in human hepatoma cells. Nucleic Acids Res. 2015, 43, 10673–10688. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Beltran, E.; Denisenko, T.V.; Zhivotovsky, B.; Bozhkov, P.V. Tudor staphylococcal nuclease: Biochemistry and functions. Cell Death Differ. 2016, 23, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Mornon, J.P. The human EBNA-2 coactivator p100: Multidomain organization and relationship to the staphylococcal nuclease fold and to the tudor protein involved in Drosophila melanogaster development. Biochem. J. 1997, 321 Pt 1, 125–132. [Google Scholar] [CrossRef]
- Shaw, N.; Zhao, M.; Cheng, C.; Xu, H.; Saarikettu, J.; Li, Y.; Da, Y.; Yao, Z.; Silvennoinen, O.; Yang, J.; et al. The multifunctional human p100 protein ‘hooks’ methylated ligands. Nat. Struct. Mol. Biol. 2007, 14, 779–784. [Google Scholar] [CrossRef]
- Friberg, A.; Corsini, L.; Mourao, A.; Sattler, M. Structure and ligand binding of the extended Tudor domain of D. melanogaster Tudor-SN. J. Mol. Biol. 2009, 387, 921–934. [Google Scholar] [CrossRef]
- Liu, K.; Chen, C.; Guo, Y.; Lam, R.; Bian, C.; Xu, C.; Zhao, D.Y.; Jin, J.; MacKenzie, F.; Pawson, T.; et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain. Proc. Natl. Acad. Sci. USA 2010, 107, 18398–18403. [Google Scholar] [CrossRef]
- Tong, X.; Drapkin, R.; Yalamanchili, R.; Mosialos, G.; Kieff, E. The Epstein-Barr virus nuclear protein 2 acidic domain forms a complex with a novel cellular coactivator that can interact with TFIIE. Mol. Cell. Biol. 1995, 15, 4735–4744. [Google Scholar] [CrossRef]
- Yang, J.; Aittomaki, S.; Pesu, M.; Carter, K.; Saarinen, J.; Kalkkinen, N.; Kieff, E.; Silvennoinen, O. Identification of p100 as a coactivator for STAT6 that bridges STAT6 with RNA polymerase II. EMBO J. 2002, 21, 4950–4958. [Google Scholar] [CrossRef]
- Paukku, K.; Yang, J.; Silvennoinen, O. Tudor and nuclease-like domains containing protein p100 function as coactivators for signal transducer and activator of transcription 5. Mol. Endocrinol. 2003, 17, 1805–1814. [Google Scholar] [CrossRef]
- Leverson, J.D.; Koskinen, P.J.; Orrico, F.C.; Rainio, E.M.; Jalkanen, K.J.; Dash, A.B.; Eisenman, R.N.; Ness, S.A. Pim-1 kinase and p100 cooperate to enhance c-Myb activity. Mol. Cell 1998, 2, 417–425. [Google Scholar] [CrossRef]
- Roworth, A.P.; Carr, S.M.; Liu, G.; Barczak, W.; Miller, R.L.; Munro, S.; Kanapin, A.; Samsonova, A.; La Thangue, N.B. Arginine methylation expands the regulatory mechanisms and extends the genomic landscape under E2F control. Sci. Adv. 2019, 5, eaaw4640. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhang, C.; Tecle, A.; Fu, X.; He, J.; Song, J.; Zhang, W.; Sun, X.; Ren, Y.; Silvennoinen, O.; et al. Tudor staphylococcal nuclease (Tudor-SN), a novel regulator facilitating G1/S phase transition, acting as a co-activator of E2F-1 in cell cycle regulation. J. Biol. Chem. 2015, 290, 7208–7220. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zhao, X.; Fu, X.; Su, C.; Xin, L.; Saarikettu, J.; Yang, X.; Yao, Z.; Silvennoinen, O.; Wei, M.; et al. Tudor-SN, a novel coactivator of peroxisome proliferator-activated receptor gamma protein, is essential for adipogenesis. J. Biol. Chem. 2014, 289, 8364–8374. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, X.; Zhu, Y.; He, J.; Shao, J.; Su, C.; Zhang, Y.; Zhang, W.; Saarikettu, J.; Silvennoinen, O.; et al. Tudor staphylococcal nuclease (Tudor-SN) participates in small ribonucleoprotein (snRNP) assembly via interacting with symmetrically dimethylated Sm proteins. J. Biol. Chem. 2012, 287, 18130–18141. [Google Scholar] [CrossRef]
- Cappellari, M.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Saarikettu, J.; Silvennoinen, O.; Sette, C. The transcriptional co-activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 2014, 33, 3794–3802. [Google Scholar] [CrossRef]
- Yang, J.; Valineva, T.; Hong, J.; Bu, T.; Yao, Z.; Jensen, O.N.; Frilander, M.J.; Silvennoinen, O. Transcriptional co-activator protein p100 interacts with snRNP proteins and facilitates the assembly of the spliceosome. Nucleic Acids Res. 2007, 35, 4485–4494. [Google Scholar] [CrossRef]
- Caudy, A.A.; Ketting, R.F.; Hammond, S.M.; Denli, A.M.; Bathoorn, A.M.; Tops, B.B.; Silva, J.M.; Myers, M.M.; Hannon, G.J.; Plasterk, R.H. A micrococcal nuclease homologue in RNAi effector complexes. Nature 2003, 425, 411–414. [Google Scholar] [CrossRef]
- Scadden, A.D. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat. Struct. Mol. Biol. 2005, 12, 489–496. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef]
- Li, C.L.; Yang, W.Z.; Chen, Y.P.; Yuan, H.S. Structural and functional insights into human Tudor-SN, a key component linking RNA interference and editing. Nucleic Acids Res. 2008, 36, 3579–3589. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Miyoshi, K.; Myers, J.R.; Du, P.; Ashton, J.M.; Tian, B.; Maquat, L.E. Tudor-SN-mediated endonucleolytic decay of human cell microRNAs promotes G1/S phase transition. Science 2017, 356, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, R.A.; Miyoshi, K.; Hedaya, O.; Myers, J.R.; Maquat, L.E. UPF1 helicase promotes TSN-mediated miRNA decay. Genes Dev. 2017, 31, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.W.; Winter, S.; Rackwitz, H.R.; Heid, H.W. Nuclear coactivator protein p100 is present in endoplasmic reticulum and lipid droplets of milk secreting cells. Biochim. Biophys. Acta 2000, 1523, 84–90. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Makarova, J.A.; Shkurnikov, M.U.; Wicklein, D.; Lange, T.; Samatov, T.R.; Turchinovich, A.A.; Tonevitsky, A.G. Intracellular and extracellular microRNA: An update on localization and biological role. Prog. Histochem. Cytochem. 2016, 51, 33–49. [Google Scholar] [CrossRef]
- Nakamura, K.; Sawada, K.; Yoshimura, A.; Kinose, Y.; Nakatsuka, E.; Kimura, T. Clinical relevance of circulating cell-free microRNAs in ovarian cancer. Mol. Cancer 2016, 15, 48. [Google Scholar] [CrossRef]
- Jelonek, K.; Wojakowska, A.; Marczak, L.; Muer, A.; Tinhofer-Keilholz, I.; Lysek-Gladysinska, M.; Widlak, P.; Pietrowska, M. Ionizing radiation affects protein composition of exosomes secreted in vitro from head and neck squamous cell carcinoma. Acta Biochim. Pol. 2015, 62, 265–272. [Google Scholar] [CrossRef]
- Silvers, C.R.; Miyamoto, H.; Messing, E.M.; Netto, G.J.; Lee, Y.F. Characterization of urinary extracellular vesicle proteins in muscle-invasive bladder cancer. Oncotarget 2017, 8, 91199–91208. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, L.; Lv, D.; Zhu, X.; Tang, H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. J. Hematol. Oncol. 2019, 12, 53. [Google Scholar] [CrossRef]
- Fashe, T.; Saarikettu, J.; Isomaki, P.; Yang, J.; Silvennoinen, O. Expression analysis of Tudor-SN protein in mouse tissues. Tissue Cell 2013, 45, 21–31. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, C.; Meng, H.; Zhang, K.; Shi, L.; Cao, C.; Wang, Y.; Su, C.; Xin, L.; Ren, Y.; et al. Oncoprotein Tudor-SN is a key determinant providing survival advantage under DNA damaging stress. Cell Death Differ. 2018, 25, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Di Lorenzo, A.; Lin, K.; Lu, Y.; Zhong, Y.; Sebastian, M.M.; Muller, W.J.; Yang, Y.; Bedford, M.T. Mouse Models of Overexpression Reveal Distinct Oncogenic Roles for Different Type I Protein Arginine Methyltransferases. Cancer Res. 2019, 79, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Cui, X.; Zhuo, Y.; Li, H.; Ha, C.; Xin, L.; Ren, Y.; Zhang, W.; Sun, X.; et al. Oncoprotein SND1 hijacks nascent MHC-I heavy chain to ER-associated degradation, leading to impaired CD8+ T cell response in tumor. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, X.; Liu, M.; Zhao, C.; Zhang, N.; Ren, Y.; Su, C.; Zhang, W.; Sun, X.; He, J.; et al. A pan-cancer analysis of the oncogenic role of staphylococcal nuclease domain-containing protein 1 (SND1) in human tumors. Genomics 2020. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Ochiai, M.; Nakashima, K.; Ubagai, T.; Sugimura, T.; Nakagama, H. SND1, a component of RNA-induced silencing complex, is up-regulated in human colon cancers and implicated in early stage colon carcinogenesis. Cancer Res. 2007, 67, 9568–9576. [Google Scholar] [CrossRef]
- Yoo, B.K.; Santhekadur, P.K.; Gredler, R.; Chen, D.; Emdad, L.; Bhutia, S.; Pannell, L.; Fisher, P.B.; Sarkar, D. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology 2011, 53, 1538–1548. [Google Scholar] [CrossRef]
- Petrick, J.L.; Kelly, S.P.; Altekruse, S.F.; McGlynn, K.A.; Rosenberg, P.S. Future of Hepatocellular Carcinoma Incidence in the United States Forecast Through 2030. J. Clin. Oncol. 2016, 34, 1787–1794. [Google Scholar] [CrossRef]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Mohamed, A.M.T.; Kolonin, M.G.; Sladek, F.M.; Younes, M.; Eckel-Mahan, K.L. HNF4alpha-Deficient Fatty Liver Provides a Permissive Environment for Sex-Independent Hepatocellular Carcinoma. Cancer Res. 2019, 79, 5860–5873. [Google Scholar] [CrossRef]
- Seyda Seydel, G.; Kucukoglu, O.; Altinbasv, A.; Demir, O.O.; Yilmaz, S.; Akkiz, H.; Otan, E.; Sowa, J.P.; Canbay, A. Economic growth leads to increase of obesity and associated hepatocellular carcinoma in developing countries. Ann. Hepatol. 2016, 15, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Hasanin, M.; Kaif, M.; Wiesner, R.; Kuo, Y.F. Nonalcoholic Steatohepatitis is the Most Rapidly Growing Indication for Simultaneous Liver Kidney Transplantation in the United States. Transplantation 2016, 100, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Tarao, K.; Tanaka, K.; Nozaki, A.; Sato, A.; Ishii, T.; Komatsu, H.; Ikeda, T.; Komatsu, T.; Matsushima, S.; Oshige, K. Efficacy and safety of dual therapy with daclatasvir and asunaprevir in elderly patients. World J. Hepatol. 2017, 9, 544–550. [Google Scholar] [CrossRef]
- Gu, J.; Yao, M.; Yao, D.; Wang, L.; Yang, X.; Yao, D. Nonalcoholic Lipid Accumulation and Hepatocyte Malignant Transformation. J. Clin. Transl. Hepatol. 2016, 4, 123–130. [Google Scholar] [CrossRef][Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Twaddel, W.; Goloubeva, O.G.; Wong, K.K.; Saxena, N.K.; Biswal, S.; Girnun, G.D. Metformin prevents liver tumorigenesis by inhibiting pathways driving hepatic lipogenesis. Cancer Prev. Res. 2012, 5, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.J.; Voorneveld, P.W.; Kodach, L.L.; Hardwick, J.C. Cholesterol metabolism and colorectal cancers. Curr. Opin. Pharmacol. 2012, 12, 690–695. [Google Scholar] [CrossRef]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of Hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef]
- Lin, Z.; Jia, H.; Hong, L.; Zheng, Y.; Shao, W.; Ren, X.; Zhu, W.; Lu, L.; Lu, M.; Zhang, J.; et al. Prognostic impact of SET domain-containing protein 8 and protein arginine methyltransferase 5 in patients with hepatocellular carcinoma following curative resection. Oncol. Lett. 2018, 16, 3665–3673. [Google Scholar] [CrossRef]
- Yin, C.; Lin, Y.; Zhang, X.; Chen, Y.X.; Zeng, X.; Yue, H.Y.; Hou, J.L.; Deng, X.; Zhang, J.P.; Han, Z.G.; et al. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology 2008, 48, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.F.; Ding, J.; Yin, C.; Zhong, W.; Wu, K.; Zeng, X.; Yang, W.; Chen, Y.X.; Zhang, J.P.; Zhang, X.; et al. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res. 2010, 70, 7640–7651. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, X.; Zhao, L.; Li, J.; Yang, H.; Zhu, Z.; Liu, J.; Huang, G. Arginine Methylation of SREBP1a via PRMT5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res. 2016, 76, 1260–1272. [Google Scholar] [CrossRef]
- Farra, R.; Grassi, G.; Tonon, F.; Abrami, M.; Grassi, M.; Pozzato, G.; Fiotti, N.; Forte, G.; Dapas, B. The Role of the Transcription Factor E2F1 in Hepatocellular Carcinoma. Curr. Drug Deliv. 2017, 14, 272–281. [Google Scholar] [CrossRef]
- Paukku, K.; Kalkkinen, N.; Silvennoinen, O.; Kontula, K.K.; Lehtonen, J.Y. p100 increases AT1R expression through interaction with AT1R 3’-UTR. Nucleic Acids Res. 2008, 36, 4474–4487. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, H.; Gow, W.; Ma, L.; Jin, Y.; Hui, B.; Yang, Z.; Wang, Z. Potential biomarkers Ang II/AT1R and S1P/S1PR1 predict the prognosis of hepatocellular carcinoma. Oncol. Lett. 2020, 20, 208. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Akiel, M.; Emdad, L.; Gredler, R.; Srivastava, J.; Rajasekaran, D.; Robertson, C.L.; Mukhopadhyay, N.D.; Fisher, P.B.; Sarkar, D. Staphylococcal nuclease domain containing-1 (SND1) promotes migration and invasion via angiotensin II type 1 receptor (AT1R) and TGFbeta signaling. FEBS Open Bio 2014, 4, 353–361. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Yu, L.; Liu, X.; Cui, K.; Di, Y.; Xin, L.; Sun, X.; Zhang, W.; Yang, X.; Wei, M.; Yao, Z.; et al. SND1 Acts Downstream of TGFbeta1 and Upstream of Smurf1 to Promote Breast Cancer Metastasis. Cancer Res. 2015, 75, 1275–1286. [Google Scholar] [CrossRef]
- Ochoa, B.; Chico, Y.; Martinez, M.J. Insights into SND1 Oncogene Promoter Regulation. Front. Oncol. 2018, 8, 606. [Google Scholar] [CrossRef]
- Bisteau, X.; Caldez, M.J.; Kaldis, P. The Complex Relationship between Liver Cancer and the Cell Cycle: A Story of Multiple Regulations. Cancers 2014, 6, 79–111. [Google Scholar] [CrossRef] [PubMed]
- Caselmann, W.H. Pathogenesis of hepatocellular carcinoma. Digestion 1998, 59 (Suppl. 2), 60–63. [Google Scholar] [CrossRef]
- Mauad, T.H.; van Nieuwkerk, C.M.; Dingemans, K.P.; Smit, J.J.; Schinkel, A.H.; Notenboom, R.G.; van den Bergh Weerman, M.A.; Verkruisen, R.P.; Groen, A.K.; Oude Elferink, R.P.; et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 1994, 145, 1237–1245. [Google Scholar] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, G.; Srivastava, N.; Goyal, M.; Rakha, E.; Lothion-Roy, J.; Mongan, N.P.; Miftakhova, R.R.; Khaiboullina, S.F.; Rizvanov, A.A.; Baranwal, M. Metadherin: A Therapeutic Target in Multiple Cancers. Front. Oncol. 2019, 9, 349. [Google Scholar] [CrossRef] [PubMed]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef]
- Song, M.S.; Grabocka, E. Stress Granules in Cancer. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Gao, X.; Ge, L.; Shao, J.; Su, C.; Zhao, H.; Saarikettu, J.; Yao, X.; Yao, Z.; Silvennoinen, O.; Yang, J. Tudor-SN interacts with and co-localizes with G3BP in stress granules under stress conditions. FEBS Lett. 2010, 584, 3525–3532. [Google Scholar] [CrossRef]
- Weissbach, R.; Scadden, A.D. Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA 2012, 18, 462–471. [Google Scholar] [CrossRef]
- Su, C.; Gao, X.; Yang, W.; Zhao, Y.; Fu, X.; Cui, X.; Zhang, C.; Xin, L.; Ren, Y.; Li, L.; et al. Phosphorylation of Tudor-SN, a novel substrate of JNK, is involved in the efficient recruitment of Tudor-SN into stress granules. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhao, C.; Yao, X.; Qian, B.; Su, C.; Ren, Y.; Yao, Z.; Gao, X.; Yang, J. SND1 acts as an anti-apoptotic factor via regulating the expression of lncRNA UCA1 in hepatocellular carcinoma. RNA Biol. 2018, 15, 1364–1375. [Google Scholar] [CrossRef]
- Yao, F.; Wang, Q.; Wu, Q. The prognostic value and mechanisms of lncRNA UCA1 in human cancer. Cancer Manag. Res. 2019, 11, 7685–7696. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arcos, I.; Rueda, Y.; Gonzalez-Kother, P.; Palacios, L.; Ochoa, B.; Fresnedo, O. Association of SND1 protein to low density lipid droplets in liver steatosis. J. Physiol. Biochem. 2010, 66, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Palacios, L.; Ochoa, B.; Gomez-Lechon, M.J.; Castell, J.V.; Fresnedo, O. Overexpression of SND p102, a rat homologue of p100 coactivator, promotes the secretion of lipoprotein phospholipids in primary hepatocytes. Biochim. Biophys. Acta 2006, 1761, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Imaz, H.; Chico, Y.; Rueda, Y.; Fresnedo, O. Channeling of newly synthesized fatty acids to cholesterol esterification limits triglyceride synthesis in SND1-overexpressing hepatoma cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 137–146. [Google Scholar] [CrossRef]
- Navarro-Imaz, H.; Ochoa, B.; Garcia-Arcos, I.; Martinez, M.J.; Chico, Y.; Fresnedo, O.; Rueda, Y. Molecular and cellular insights into the role of SND1 in lipid metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158589. [Google Scholar] [CrossRef]
- Shen, Y.; Gao, G.; Yu, X.; Kim, H.; Wang, L.; Xie, L.; Schwarz, M.; Chen, X.; Guccione, E.; Liu, J.; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem. 2020, 63, 9977–9989. [Google Scholar] [CrossRef]
- Palte, R.L.; Schneider, S.E.; Altman, M.D.; Hayes, R.P.; Kawamura, S.; Lacey, B.M.; Mansueto, M.S.; Reutershan, M.; Siliphaivanh, P.; Sondey, C.; et al. Allosteric Modulation of Protein Arginine Methyltransferase 5 (PRMT5). ACS Med. Chem. Lett 2020, 11, 1688–1693. [Google Scholar] [CrossRef]
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.P.; Montes de Oca, R.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B.; et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018, 8, 9711. [Google Scholar] [CrossRef]
- Metz, P.J.; Ching, K.A.; Xie, T.; Delgado Cuenca, P.; Niessen, S.; Tatlock, J.H.; Jensen-Pergakes, K.; Murray, B.W. Symmetric Arginine Dimethylation Is Selectively Required for mRNA Splicing and the Initiation of Type I and Type III Interferon Signaling. Cell Rep. 2020, 30, 1935–1950. [Google Scholar] [CrossRef] [PubMed]
- Snyder, K.J.; Zitzer, N.C.; Gao, Y.; Choe, H.K.; Sell, N.E.; Neidemire-Colley, L.; Ignaci, A.; Kale, C.; Devine, R.D.; Abad, M.G.; et al. PRMT5 regulates T cell interferon response and is a target for acute graft-versus-host disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, M.; Zhang, Y.W.; Tong, S.; Leal, R.A.; Shetty, R.; Vaddi, K.; Luengo, J.I. Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1033–1038. [Google Scholar] [CrossRef]
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.; Kennedy, S.; Li, B.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. [Google Scholar] [CrossRef]
- Barczak, W.; Jin, L.; Carr, S.M.; Munro, S.; Ward, S.; Kanapin, A.; Samsonova, A.; La Thangue, N.B. PRMT5 promotes cancer cell migration and invasion through the E2F pathway. Cell Death Dis. 2020, 11, 572. [Google Scholar] [CrossRef]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, L.; Villarreal, O.D.; He, W.; Su, D.; Bedford, E.; Moh, P.; Shen, J.; Shi, X.; Bedford, M.T.; et al. PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res. 2019, 47, 5038–5048. [Google Scholar] [CrossRef] [PubMed]
- Drew, A.E.; Moradei, O.; Jacques, S.L.; Rioux, N.; Boriack-Sjodin, A.P.; Allain, C.; Scott, M.P.; Jin, L.; Raimondi, A.; Handler, J.L.; et al. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci. Rep. 2017, 7, 17993. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Company | Compound | Trials | MOA | Ref. or Patent |
|---|---|---|---|---|
| GSK/Epizyme | GSK3326595 | Ph 1 NCT02783300Ph 2 NCT03614728 | b | [164] |
| Pfizer | PF-06939999 | Ph 1 NCT03854227 | a | [165] |
| Janssen | JNJ64619178 | Ph 1 NCT03573310 | c | AACR |
| Prelude | PRT543 | Ph 1 NCT03886831 | a | [166] |
| Prelude | PRT811 | Ph 1 NCT04089449 | a | [166] |
| Prelude | C449 | Pre-clinical | f | [167] |
| Eli Lilly | LLY-283 | Pre-clinical | a | [168] |
| Argonaut | T1-44 | Pre-clinical | b | [169] |
| Jian Jin | MS4322 | Pre-clinical | d | [159] |
| Merck | Comp 1A | Pre-clinical | e | [160] |
| Aurigene | AU-14755 | Pre-clinical | b | WO2019-180628 |
| Angex | Not named | Pre-clinical | a | WO2019-112719 |
| Jubilant | JPRMT5i | Pre-clinical | b | WO2019-102494 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, T.; Wang, Y.; Bedford, M.T. The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma. Epigenomes 2021, 5, 2. https://doi.org/10.3390/epigenomes5010002
Wright T, Wang Y, Bedford MT. The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma. Epigenomes. 2021; 5(1):2. https://doi.org/10.3390/epigenomes5010002
Chicago/Turabian StyleWright, Tanner, Yalong Wang, and Mark T. Bedford. 2021. "The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma" Epigenomes 5, no. 1: 2. https://doi.org/10.3390/epigenomes5010002
APA StyleWright, T., Wang, Y., & Bedford, M. T. (2021). The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma. Epigenomes, 5(1), 2. https://doi.org/10.3390/epigenomes5010002