Abstract
EMT (epithelial to mesenchymal transition) is a plastic phenomenon involved in metastasis formation. Its plasticity is conferred in a great part by its epigenetic regulation. It has been reported that the trimethylation of lysine 27 histone H3 (H3K27me3) was a master regulator of EMT through two antagonist enzymes that regulate this mark, the methyltransferase EZH2 (enhancer of zeste homolog 2) and the lysine demethylase KDM6B (lysine femethylase 6B). Here we report that EZH2 and KDM6B are overexpressed in numerous cancers and involved in the aggressive phenotype and EMT in various cell lines by regulating a specific subset of genes. The first paradoxical role of these enzymes is that they are antagonistic, but both involved in cancer aggressiveness and EMT. The second paradoxical role of EZH2 and KDM6B during EMT and cancer aggressiveness is that they are also inactivated or under-expressed in some cancer types and linked to epithelial phenotypes in other cancer cell lines. We also report that new cancer therapeutic strategies are targeting KDM6B and EZH2, but the specificity of these treatments may be increased by learning more about the mechanisms of action of these enzymes and their specific partners or target genes in different cancer types.
1. Introduction
1.1. Epithelial to Mesenchymal Transition (EMT)
Epithelial to mesenchymal transition (EMT) is a phenomenon that leads epithelial cells to gradually acquire a mesenchymal phenotype. This process is associated with a loss of expression of specific epithelial proteins called “epithelial markers,” such as E-CADHERIN (coded by CDH1 gene), CLAUDIN, or OCCLUDIN, and a gain of expression of other proteins called “mesenchymal markers,” such as VIMENTIN or N-CADHERIN. At least three distinct types of EMT have been reported so far, even if they present highly similar mechanisms: (i) type I is implicated in embryo formation and organ development; (ii) type II is involved in wound healing, regeneration of tissues, and in organ fibrosis; and (iii) type III is implicated in cancer development and aggressiveness. The latter also plays a role in tumor escape, impairing the formation of the immune synapse or leading to the overexpression of the immune checkpoint inhibitor PD-L1 (programmed death ligand 1) [1,2,3], and in metastasis formation [4,5]. Indeed, in type III EMT, tumor cells have been shown to present a loss of expression of adhesion proteins [6], to secrete various metalloproteases, such as MMP9 or ADAM19 [7], leading to the digestion of the extracellular matrix and then to the escape of the primary tumor and the migration through the blood or lymphatic circulation [4,5].
All the changes occurring during EMT at the morphological, functional, or molecular levels can be reversed to return to the original epithelial phenotype. This phenomenon is called mesenchymal to epithelial transition (MET) [8,9]. Through this process, circulating cancer cells can invade another organ, leading to the formation and growth of a secondary tumor called metastasis (Figure 1). However, the definition of EMT remains complex since there is not only one EMT but a multitude of intermediate states presenting different expressions of epithelial and mesenchymal markers. This plasticity has been described to be mainly linked to the epigenetic regulation of EMT, which is itself a highly plastic and reversible process, as described below.
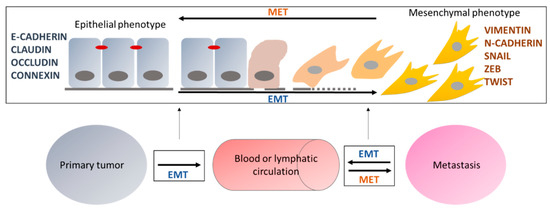
Figure 1.
Roles of EMT in the formation of metastases. Epithelial cells, expressing epithelial markers, can undergo EMT and progressively acquire a mesenchymal phenotype characterized by the expression of mesenchymal markers. EMT is reversible and the opposite transition is called MET (mesenchymal to epithelial transition). EMT and MET are involved in the emergence of metastases by allowing cells to migrate to the blood or lymphatic circulation and then invade another organ to generate a secondary tumor called metastasis.
Type III EMT can be induced by various intra- or extra-cellular factors or stress. It has been described that EMT-activating transcription factors (EMT-ATFs), such as SNAIL, TWIST, and ZEB (zinc finger E-box-binding homeobox) families, are main regulators of EMT [10]. These EMT-ATFs are activated via hypoxia, cytokines (TGFβ, TNFα), growth factors (EGF), or by cellular signaling pathways (WNT, HEDGEHOG, NF-κB, and NOTCH), and their activation leads to EMT induction [11].
1.2. Epigenetic Regulation of EMT
The word “epigenetics” was used for the first time in 1942 by Conrad Hal Waddington, who used the term of “epigenotype” to describe the different phenomena which are involved in the implementation of a phenotype from a genotype, and “epigenetics” for studies related to the understanding of this phenomenon [12]. This definition has been widely modified since the 1940s to lead to the one currently used now: “Epigenetics is the study of the regulation of gene expression, without DNA nucleotide sequence alteration, and linked to hereditary transmission.” This field includes the study of non-coding RNA (miRNA, lncRNA, etc.), which can bind DNA or mRNA and influence gene transcription, DNA methylation (methylation of cytosines in CpG regions leading to the repression of transcription) and histone post-translational modifications (Table 1). Plasticity of epigenetic regulation is conferred by the reversible character of the corresponding biochemical reactions. Indeed, DNA methylation, histone post-translational modifications, and non-coding RNA expression can be reversed according to the cell pathophysiological context. Moreover, epigenetic modifications have been widely linked to EMT regulation and suggested to confer the necessary plasticity to regulate cancer aggressiveness, invasion, and metastasis [13].

Table 1.
Main histone post-translational modifications and their effects on gene expression.
In this review, we will focus on the regulation of the methylation of lysine 27 of histone H3 (H3K27me3), a repressive mark, which has been described as a master regulator of EMT. It is noteworthy that the amino group of lysine 27 of histone H3 can also be acetylated and that the H3K27Ac mark is generally associated with increased gene transcription and therefore inversely correlated with H3K27me3 [14,15]. It has also been shown that the H3K27me3 mark is associated with temporarily repressed gene expression, which could easily be re-expressed later [16]. H3K27 can be mono-, di-, or tri-methylated (H3K27me, H3K27me2, and H3K27me3). H3K27me is associated with active transcription, whereas H3K27me2, and in particular H3K27me3, are associated with repressed genes (Figure 2). This methylation status is regulated via a histone methyltransferase, EZH2 (enhancer of zeste homolog 2), which mono-, di-, or tri-methylates H3K27; and two lysine demethylases, KDM6B (lysine demethylase 6B) and KDM6A (lysine demethylase 6A), which can mono-, di-, or tri-demethylate H3K27.
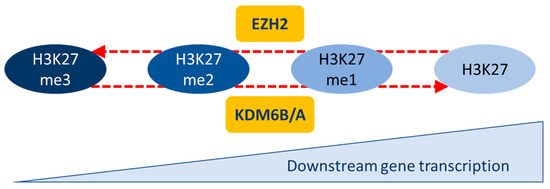
Figure 2.
Regulation of H3K27 methylation status. H3K27 methylation status is regulated via three enzymes: EZH2 (methyltransferase) and KDM6B/A (demethylases). The H3K27 methylation level is inversely correlated to downstream gene transcription.
1.3. Current Described Roles of KDM6B and EZH2
EZH2, also called KMT6A (lysine methyl-transferase 6A or ENX-1), is a subunit of the polycomb repressive complex 2 (PRC2), which is normally involved in embryonic development and in stem cell maintenance. PRC2’s main function is the di- or tri-methylation of H3K27, via its catalytic subunit EZH2. The other essential components of the PRC2 complex are SUZ12 (suppressor of zeste 12) and EED (embryonic ectoderm development). SUZ12 is directly involved in the tri-methylation of H3K27me3, more likely by inducing EZH2 binding to histone and modulating EZH2 conformation [17]. EED has a H3K27 binding domain that favors the recruitment of PRC2 on histone H3. Additional proteins have also been associated with the PRC2 complex: RBBP4 and RBBP7 (retinoblastoma binding protein 4 and 7), and HDAC1 and HDAC2 (histone deacetylase 1 and 2). However, these proteins are not required for the histone methyltransferase activity of PRC2 [18] but they may drive the recruitment of the PRC2 complex onto DNA or histone H3 (Figure 3). PRC2 can regulate different subsets of genes depending on the cell type. Indeed, it has been described that PRC2 may be recruited to chromatin on a response element (PRE = PRC2 response element) [19], but the cell type-dependent profiles of PRC2 recruitment remain unclear. However, the potential presence of specific DNA- (or histone)-binding proteins in the PRC2 complex (e.g., HDAC1 and 2 or RBBP4 and 7) may partially explain these cell type specificities (Figure 3).
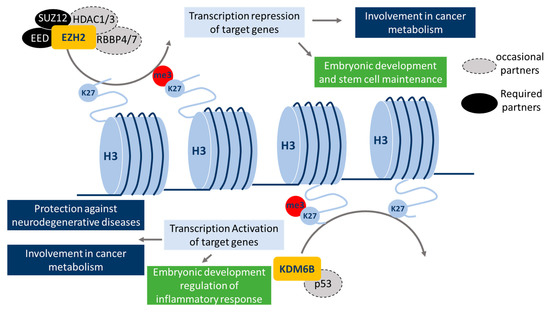
Figure 3.
The current described roles of EZH2 and KDM6B. EZH2 methyltransferase activity, which catalyzes mono-, di-, or tri-methylation of H3K27, requires the presence of the PRC2-associated proteins, SUZ12 and EED. HDAC 1/3 and RBBP 4/7 can also be part of this complex but are not required for methyltransferase activity. EZH2 is important for normal embryonic development and stem cell maintenance. Moreover, it can also regulate cancer metabolism. KDM6B presents a lysine demethylase activity, which is a specific feature of H3K27. p53 has been reported to interact with KDM6B but is not required for its demethylase activity. KDM6B is required for embryonic development and the regulation of inflammatory responses, and is also important in cancer metabolism regulation and presents protective effects in some neurodegenerative diseases.
KDM6B, also called JMJD3 (Jumonji domain containing protein 3), is a H3K27me3 lysine demethylase, which plays an important role in the regulation of embryogenic development via controlling HOX genes (a subset of homeotic genes). For example, KDM6B can directly demethylate H3K27me3 in HOX and BMP (bone morphogenetic protein) promoters leading to activated transcription, which is essential for bone differentiation [20]. KDM6B also plays a role in the regulation of the inflammatory response by controlling macrophage differentiation (transcription activation of HOXA11 and BMP2 genes) [21]. The proteins involved in the recruitment of KDM6B on chromatin are poorly documented. Nonetheless, it has been shown that p53 can bind to KDM6B and promotes the recruitment of the complex on its target genes to activate their transcription [22].
Furthermore, even if PRC2 can bind to its PRE, this is not sufficient to explain the target genes of EZH2, and KDM6B appears to not have any affinity to specific DNA regions. EZH2 and KDM6B may rather require protein partners to specifically bind chromatin (Figure 3).
The deregulation of KDM6B and EZH2 has been described to be involved in the development of several disorders. KDM6B seems to prevent some neurodegenerative diseases, such as Alzheimer’s disease [23], but the most documented disease linked to KDM6B and EZH2 dysregulation is cancer [23,24,25]. Indeed, both these enzymes have been involved in EMT and tumor aggressiveness in various cancer cell types, and this review will specifically focus on the roles of EZH2 and KDM6B on the epigenetic regulation of EMT. Despite their described antagonist roles, these two enzymes were almost never studied together, apart from the work from Daures et al. [26,27].
2. EZH2 Is Associated to EMT and Cancer Aggressiveness
2.1. EZH2 Is Differently Expressed between Tumor and Normal Tissues in Various Cancer Types
A great number of publications studied EZH2 expression in different tumors using various techniques. These data have been summarized in Table 2. In clear cell renal cell carcinomas (ccRCCs), a high expression of EZH2 has been correlated with bad prognosis, metastasis, and high grade tumors [28,29]. Similar observations were collected in esophagus squamous cell carcinoma, in which EZH2 expression levels are higher in tumors compared to adjacent tissues and this higher expression has been correlated to the apparition of distant metastasis, bad prognosis, depth of invasion, and tumor size [30]. Similar data were also obtained in bladder urothelial carcinoma; in cutaneous (human and murine) melanoma; in hormone refractory and metastatic prostate cancer; in gastric cancer; breast, lung, and ovarian carcinoma; as well as in pediatric brain tumors. Therefore, it is clear that a high expression of EZH2 was frequently correlated to high grade tumors, aggressiveness, and poor patient outcome [31,32,33,34,35,36,37,38,39,40].
Table 2.
EZH2 expression in various cancer types and correlation to clinical parameters. EZH2 is often overexpressed in cancers (green), but in a few cases such as lung adenocarcinoma or myelodysplastic syndromes, some inactivating mutations (red) have been found in the EZH2 gene. IHC: immunohistochemistry; IF: immunofluorescence.
Although EZH2 seems generally associated to aggressiveness, some inactivating mutations or deletions of the EZH2 gene have also been reported in different cancers, suggesting an ambiguous role of EZH2 in these cancers. In lung adenocarcinomas, the KRAS mutation is a major oncogenic mutation frequently associated with the EZH2 mutation. Inactivating EZH2 mutations was found in 14% of all the studied tumors. Conditional EZH2 knock-out (KO) mice were also used to demonstrate that EZH2 inactivation increased lung adenocarcinoma aggressiveness in mice carrying the KRAS mutation [41]. Moreover, a study performed on 119 patients with myelodysplastic syndromes and myeloproliferative neoplasms showed that 8.4% of the patients presented an inactivating mutation in the EZH2 gene and that 3.4% of patients presented a deletion of this gene. Moreover, the deletion of the EZH2 gene in mice was sufficient to induce a myeloid dysplasia [42].
2.2. EZH2 Modulates EMT by Targeting a Specific Subset of Genes
Since EZH2 was described to be overexpressed in many aggressive tumors, this protein was proposed to act as a master regulator of the expression of EMT ATFs. However, it has been shown, in head and neck squamous cell carcinoma cell lines FaDu and SNU1041, that EZH2 did not regulate SNAI1 or SNAI2 gene expression. Moreover, a siRNA-driven knock-down of EZH2 increased E-CADHERIN levels and decreased N-CADHERIN and VIMENTIN levels, leading to an inhibition of migration and invasion capacities without modulating EMT ATFs levels [43]. Surprisingly, in the ovarian cancer cells SKOV3, ChIP experiments showed that EZH2 could directly repress ZEB2 expression when EMT was induced in these cells following TGF-β treatment [44] suggesting that EZH2 inhibited EMT in these cells. In the MHCC97H and MHCC97L cells lines (hepatocellular carcinomas), lncRNA Carlo5 bound to EZH2 protein and recruited it onto the miR-200b promoter to repress its transcription. miR-200b was previously described to be linked to an epithelial phenotype by blocking EMT, provoked by the inhibition of ZEB1 and ZEB2 signaling (repressors of CDH1 gene transcription). The repression of miR-200b expression by the complex lncRNA Carlo5-EZH2 therefore favored EMT [45]. In non-small cell lung cancer cells, miR-21 overexpression also enhanced EZH2 expression via an unknown mechanism and promoted cell invasion and migration [46].
Even if the implication of EZH2 in the regulation of EMT ATFs expression is not clearly defined, many interactions between these proteins have been reported. Indeed, overexpression of TWIST1 in bone marrow squamous cells led to the overexpression of EZH2 via an unknown mechanism. This was also associated with the repression of cell proliferation mediated by the recruitment of EZH2 and addition of the H3K27me3 marks on the P16/INK4/ARF locus [47]. In oral tongue squamous cell carcinomas cells, Cal27, SNAI1, and SNAI2 induced the repression of miR-101, and consequently increased the levels of EZH2 in an unclear mechanism [48]. In the nasopharyngeal carcinoma cell line, HNE1 and SNAI1 can bind to the deacetylase enzymes HDAC1/2, which has also been described to interact with EZH2. This linear repressive complex is then able to trimethylate H3K27 and deacetylate histones, and thus repress the transcription of target genes, such as CDH1 [49].
EZH2 did not only regulate EMT via its interaction with EMT ATFs, but also by regulating several target genes that were described as EMT regulators in many cell types (Table 3). Indeed, the down-regulation of CDH1 can be directly controlled via EZH2. It has been shown in the colon cancer cell line HT-29-M6 that both SNAI1 and EZH2 were required to recruit PRC2 on the promoter of CDH1, and that EZH2 and SNAI1 stabilized each other on this promoter [50]. Cho and collaborators also reported that EZH2 directly repressed CDH1 expression by the trimethylation of H3K27 on its promoter [51]. Similarly, it has been suggested, in the ovarian cancer cell line HO-8910, that EZH2 repressed CDH1 gene expression. Indeed, a siRNA-driven knock-down of EZH2 led to an increase in E-CADHERIN expression and to a decrease in invasion and migration capacities of these cells [36]. In the prostate cancer cell line PC3, the chemical compound triptolide (a promising anti-cancer drug whose mechanisms are not well described but are believed to involve the regulation of caspases, NF-κB pathway, heat-shock proteins, and DNA repair associated factors [52]) decreased mRNA levels of EZH2, and led to an increase in CDH1 expression [53].
Table 3.
EZH2 target genes in various cancer cell lines. EZH2 directly downregulated (red) many genes in a wide range of cancer cell lines, but in some cases, it also directly, or indirectly, activated (green) transcription of some genes such as NF-κB target genes, C-MYC or CYCLIN D1.
RKIP (Raf-1 kinase protein inhibitor) inhibited tumor metastasis in different types of cancers such as breast, prostate, gastric, and cervical cancers [54,55,56]. In breast (MCF-7, T47-D, and MDA-MB-231) and prostate (LNCaP, DU145, and PC3) cancer cell lines, EZH2 directly repressed RKIP transcription by inducing H3K27 trimethylation on its promoter, leading to increased metastasis [57]. Furthermore, in melanomas, EZH2 similarly repressed AMD1 gene expression (a protein involved in polyamine synthesis), leading to increased levels of SNAI1, ZEB1, and TWIST1, and to increased invasion and metastasis, both in vitro and in vivo [40]. In small cell lung cancers, EZH2 overexpression is related to poor prognosis and associated to the H3K27me3-dependent repression of JUB1 in these tumors. Moreover, overexpression of JUB1 in small cell lung cancer cells—DMS53, Lul30, and H209 cells—decreased cell proliferation showing that EZH2 promoted lung cancer cell proliferation via the inhibition of JUB1 expression in these models [58]. P27kip, a gene coding a tumor suppressor protein involved in cell cycle arrest via the inhibition of CDK proteins, presented an EZH2-dependent repression of transcription in MIA-PaCa2 and Panc04.03 cancer cells, leading to uncontrolled cell growth and aggressiveness [59].
EZH2 also promoted EMT and cancer aggressiveness via the repression of genes involved in different signaling pathways. Indeed, in liver cancers, EZH2 was recruited by the long non-coding RNA lncAPC to the APC promoter where it repressed transcription in a H3K27 methylation-dependent manner. APC normally participates to the cytosolic sequestration of β-CATENIN and to its degradation. EZH2 overexpression therefore led to the activation of the Wnt/β-CATENIN pathway that favored liver tumor initiating cells (TIC) self-renewal and cancer aggressiveness [60]. In the non-small cell lung adenocarcinoma cell lines, A549 and H358, chemical inhibition (using the specific GSK126 inhibitor) of EZH2 led to increased levels of IGF1 mRNAs, associated to a global decrease in H3K27me3 levels [41]. Amongst the IGF1 downstream targets, AKT and ERK signaling pathways were previously described to be both associated to increased proliferation.
Data obtained in breast cancers also illustrated the dual role of EZH2 in cancer aggressiveness. In the MDA-MB-231 cell line, an ER (estrogen receptor)-negative cell line, EZH2 positively activated the transcription of NF-κB target genes in a methyltransferase-independent way, which contributed to aggressiveness of these breast cancer cells. On the contrary, in the ER-positive breast cancer cell line, MCF-7, EZH2 interacted with ER and activated the recruitment of the PRC2 complex on the NF-κB promoter where EZH2 tri-methylated H3K27 and repressed NF-κB transcription [61].
Altogether, these studies highlighted that EZH2 can be considered as an inducer of EMT or cancer aggressiveness in a large range of tumors, but surprisingly, in rare cases, EZH2 was also associated to anti-tumor properties. These studies highlighted the paradoxical role of EZH2 in the regulation of EMT.
2.3. EZH2 Modulates EMT and Cancer Aggressiveness via Activating Gene Transcription, Independently of Its Methyltransferase Catalytic Activity
As described above, EZH2 might present both methyltransferase and polycomb-independent activities (Table 3). In the absence of ER, in ER-negative breast cancers, EZH2 interacted with RelA and RelB, two NF-κB subunit proteins, but surprisingly, EZH2 mutants lacking the enzymatic domain were still able to interact with both these proteins showing that these interactions were independent of the catalytic subunit of EZH2. Furthermore, in cells overexpressing either an inactive or an active EZH2, without expressing wild type EZH2, the transcription of NF-κB target genes was up-regulated, showing that EZH2 activated transcription of these genes independently of its catalytic activity. Moreover, co-immunoprecipitation experiments of SUZ12 confirmed the interaction of SUZ12 with EZH2, but not with RelA or RelB, data which strongly suggested that the binding of EZH2 with the PRC2 polycomb complex or RelA/RelB was exclusive. These data therefore suggested that the Polycomb complex was not required for the transcriptional role of EZH2 [61].
Similarly, in the breast cancer cell lines, MCF-7 and T47D, co-IP and GST-pulldown experiments revealed that EZH2 was found to interact with ERα and β-CATENIN and to induce the transcription of their target genes. In these models, these interactions contributed to cell cycle progression and cancer aggressiveness [62]. These examples also strongly suggested that EZH2 was able to induce the transcription of target genes independently of its interaction with the PRC2 complex.
2.4. Regulation of EZH2 Expression during EMT
EZH2 expression was described to be regulated at both transcriptional and post-translational levels. Two different transcription factors have been reported to activate EZH2 transcription during EMT. First, in the breast epithelial cancerous and normal cells, PY2T and NMuMG, SOX4 was described as a direct activator of EZH2 transcription following TGF-β treatment [63]. Second, in the breast cancer cell lines—MDA-MB-468, MDA-MB-231, and SKBr3—the MEK-ERK1/2 pathway activated EZH2 transcription via the direct recruitment of P-ELK1 on the EZH2 promoter. Indeed, this activation was not observed in the less aggressive breast cancer cell line HCC1500 (ER-positive cells) [64].
Many miRNAs were also described to target EZH2 mRNA leading to decreased cellular levels of EZH2 mRNA. Among these miRNAs, miR-101 was the most frequently observed, since it has been described to target EZH2 in gastric cancer cell lines (BGC-823, SGC-7901, AGS, and MKN-45), in head and neck squamous cell carcinomas (HNSCCs) and in the glioblastoma cell line U87. In all these cancer models, miR-101 inhibited migration, invasion, or EMT [65,66,67]. Similarly, in the hepatocellular carcinoma cell line HEPG2, another miRNA, miR-137, targeted EZH2 mRNA that led to decreased cell migration and invasion. Indeed, miR-137 was described to be less expressed in HCCs than in normal tissues [68]. Moreover, miR-138 also inhibited EMT by targeting EZH2 mRNA in the HNSCCs 1386Ln and 686Tu cell lines [69]. miR-124 targeted the 3’-UTR of EZH2 mRNA leading to its degradation and overexpression of this miRNA suppressed cell motility, invasion and EMT in vitro, and intra-hepatic and pulmonary metastasis formation in vivo [70]. In the esophageal squamous cell carcinoma cell line, Ecal09, miR-98, and miR-214 also targeted the 3’-UTR of EZH2 mRNA and inhibited migration and invasion. These miRNAs were downregulated in esophageal squamous cell carcinomas and their expression was inversely correlated with lymph node metastasis [71]. Finally, various miRNA targeted EZH2 mRNA and inhibit migration and invasion in various cancer cell lines. MiR-214, miR-4465, and miR-137 targeted EZH2 respectively in hepatocellular carcinoma cell line SK-HEP-1, in non-small cell lung cancer cell lines A549 and H2170, and in melanoma cell lines Ma-Mel-79b and -86b [72,73,74]
Altogether, these data demonstrated that the EZH2 gene and EZH2 mRNA levels are tightly regulated by both transcription factors and miRNAs during EMT.
2.5. New Anti-Cancer Therapeutic Protocols Targeting EZH2 Activity
More than 10 clinical studies are evaluating the effects of EZH2 inhibitors (EZH2i) against multiple cancers (https://clinicaltrials.gov). For example, a phase I study using the EZH2 chemical inhibitor GSK2816126 in elapsed/refractory diffuse large B-Cell lymphoma (DLBCL), other non-Hodgkin lymphomas (NHL), transformed follicular lymphoma (tFL), solid tumors, and multiple myeloma (MM) showed an evident anti-tumoral effect of the molecule [75]. Many different EZH2i are tested such as tazemetostat (used in almost 15 studies), MK683, CPI-1205, and SHR2554. Although these compounds are still being evaluated, it appears that EZH2 inhibition is a very promising therapeutic approach in a wide range of cancer types (solid tumors or not) (Table 4).
Table 4.
Summary of the clinical trials targeting EZH2 in cancers. Different molecules targeting EZH2 are used in clinical trials against a large range of solid and liquid tumors.
3. KDM6B in EMT and Cancer Aggressiveness
3.1. KDM6B Is Differently Expressed between Tumor and Normal Tissues in Various Cancer Types
KDM6B has been described to be differentially expressed between cancer and normal adjacent tissues (Table 2). For example, in hepatocellular carcinomas, tissue-based RT-qPCR and Western-blotting analysis showed that KDM6B was overexpressed in tumor tissues compared to normal adjacent tissues. Moreover, KDM6B expression was correlated with distant metastasis and with an increased expression of SNAI2 [76]. In clear cell renal cell carcinomas, KDM6B expression was also higher in tumor tissues compared to normal ones (tissue-based RT-qPCR, Western blotting, and IHC), and overexpression of KDM6B was also linked to increased tumor size, metastasis, and poor prognosis for patients [77]. Similar results were observed in multiple myelomas according to a public data set study [78] and in Hodgkin’s lymphomas (IHC analysis) [79]. An Oncomine data analysis also confirmed that KDM6B was overexpressed in invasive and metastatic breast tumors compared to less invasive tumors [80]. Similarly, RT-PCR data showed that KDM6B mRNA was overexpressed in malignant pleural mesothelioma (MPM) compared to benign pleura [81]. Moreover, KDM6B was also overexpressed in ovarian cancers at both protein and mRNA levels and its expression was associated with invasion, metastasis, and low overall survival [82]. All these studies therefore clearly indicated that KDM6B could be classified as an oncogene and was correlated to EMT and cancer aggressiveness. However, some contradictory or intriguing results have also been reported. For example, even if KDM6B was overexpressed in pancreatic ductile adenocarcinoma (PDAC) compared to normal tissue, its expression decreased with tumor grades [83]. Moreover, KDM6B was under-expressed in poor differentiated tumors (mesenchymal phenotype) in a cohort of 96 colon cancer patients [84]. In 20 colorectal cancer patients, KDM6B was also under-expressed compared to normal tissues, and a high KDM6B expression was correlated with better overall survival in a cohort of 151 colorectal cancer (CRC) patients [85]. Altogether, these data suggested that KDM6B could be correlated, or inversely correlated, with invasion and cancer aggressiveness regarding the cancer model. All these data are summarized in Table 5.
Table 5.
KDM6B expression in different cancer types. KDM6B is often overexpressed (green) in cancers; however, it was described to be under-expressed (red) in invasive pancreatic ductal adenocarcinomas and poor differentiated colon cancers.
3.2. KDM6B Modulates EMT by Targeting a Specific Subset of Genes
KDM6B was described to modulate the expression of EMT ATFs during EMT in various cancer cell lines (Table 6). Indeed, the expression of SNAIL was directly activated by KDM6B. For example, in the mammary mouse cell line, NmuMG, H3K27 was demethylated by KDM6B on the SNAI1 promoter when KDM6B was overexpressed, leading to the expression of mesenchymal-related genes [80]. In the hepatocellular carcinoma cell line, HepG2, KDM6B directly activated SNAI2 transcription via a demethylation of H3K27 on its promoter and KDM6B overexpression promoted EMT, migration and invasion capacities and stem cell-like features (holoclone formation, clonogenic capacities, and sphere establishment) [76]. In the clear-cell renal cell carcinoma cell line, Caki-2, KDM6B also directly activated SNAI2 transcription to increase mesenchymal marker expression, decreased epithelial protein expression, and increased invasion capacities [77]. In the renal cell carcinoma cell line, ACHN, the siRNA-driven knock-down of lncRNA HOTAIR led to decreased levels of both KDM6B and its target SNAIL1, and then decreased invasion and migration capacities of the cells [86], suggesting that KDM6B was also regulated at the mRNA level.
Table 6.
KDM6B target genes in various cancer cell lines. KDM6B directly activated (green) transcription of EMT-related genes in a wide range of cancer cell lines. It also down-regulated (red) the expression of EMT-ATFs, probably via an indirect way.
On the contrary, the knock-down of KDM6B or the expression of an inactive KDM6B mutant in SW-480 human colon cancer cells increased the expression of SNAIL1, ZEB1, and ZEB2 EMT ATFs, and led to consecutive decreases in E-CADHERIN, CLAUDIN1, and CLAUDIN7 expressions (epithelial markers) [84]. These data suggested that the KDM6B demethylase activity was frequently associated to EMT-related phenotypes, but some apparent contradictory roles of this enzyme could also be dependent on the model.
KDM6B has been described to modulate the expression of various genes involved in epithelial or mesenchymal cell state. Indeed, in the colorectal carcinoma cell line, HCT116, KDM6B directly activated the expression of the epithelial protein EPCAM (epithelial cell adhesion molecule) via its demethylase activity, leading to increased tumor growth, cell proliferation, and more surprisingly, to cell migration and invasion [87]. However, it should be noted that EpCAM and WNT/β-catenin expressions were inversely correlated with survival prognosis for patients with colon cancer [88]. In ovarian cancers, HER2 (human epidermal growth factor receptor 2, a receptor involved in proliferation, migration, and survival) and KDM6B expressions were correlated to poor prognosis and a low survival rate. KDM6B promoted EMT in the SKOV-3 ovarian cancer cell line and directly activated HER2 gene transcription. Treatment of ovarian cancer cells with both Paclitaxel (which blocked the depolymerization of microtubule assembly in the mitotic spindle) and Carboplatin (a DNA alkylation chemical) decreased KDM6B content and was linked to an increase of the H3K27me3 mark on the HER2 promoter leading to its decreased expression and decreased invasive and migratory cell capacities [89]. In the breast cancer cell lines, MDA-MB-231 and MCF7, the inhibition of the KDM6B demethylase activity, using the specific inhibitor GSKJ4, led to a repression of the expression of the multipotent SOX2, NANOG, and OCT4 genes, a decrease of which was linked to an increase of the H3K27me3 mark on their promoters. These transcription factors have been described to be essential for the maintenance of the stem-like phenotype, suggesting that KDM6B regulated stemness properties of breast cancer cells [90]. On the contrary, KDM6B also promoted an epithelial-like phenotype, since, in the pancreatic ductal adenocarcinoma cell line BxPC3 and in xenograft models, KDM6B directly induced the demethylation of the H3K27me3 mark on the CEBPA promoter and induced the expression of this gene. Moreover, both knock-out of KDM6B or CEBPA led to the appearance of a mesenchymal phenotype and increased invasion capacities of these cells [83]. In the BJ foreskin cell line, KDM6B was recruited on a p53 binding element via its direct interaction with p53 where it demethylated the H3K27me3 mark. Indeed, KDM6B was proposed to participate to p53 target genes up-regulation and therefore, to cell cycle arrest or apoptosis and decreased cancer aggressiveness [22].
Some signaling pathways closely linked to EMT were also described to be regulated by KDM6B. In the melanoma A375-LM3 cell line, KDM6B demethylated H3K27 at the NF-κB and BMP promoters leading to the activation of NF-κB and BMP signaling target genes and thus contributed to the malignant cell properties. Moreover, the BMP signaling pathway activated KDM6B transcription via a positive feedback regulation loop [91]. In multiple myeloma, KDM6B was also induced via the NF-κB pathway and activated the MAPK pathway through its direct recruitment onto the ELK1 and FOS promoters leading to induced cell growth, proliferation, and aggressiveness [78]. On the contrary, in human fibroblasts, KDM6B directly activated P16/INK4A gene transcription via demethylation of H3K27 on its promoter leading to a P16-dependent arrest of the cell cycle. [92,93]. Similar results were also described in the human lung fibroblast cells, TIG-3, after induction of KDM6B expression following the BRAF pathway activation. Indeed, KDM6B was able to block cell proliferation in these cells following its recruitment on the P16/INK4A promoter [94]. All these data are summarized in Table 5.
3.3. KDM6B Modulates Cancer Aggressiveness by Activating Gene Transcription, Independently of Its Demethylase Catalytic Activity
KDM6B can also modulate cancer aggressiveness in a demethylase-independent manner. In the immortalized human embryonic kidney cell line, HEK-293, KDM6B favored the expression of p53 target genes without being recruited on their promoters, but rather through increasing p53 recruitment on its target sequences in these promoters. Indeed, overexpression of KDM6B induced the nuclear localization of p53 in these cells and the recruitment of p53 on the p21 promoter, but not of KDM6B at this site. These data were in favor of a tumor suppressor role of KDM6B via controlling the expression of p53 downstream genes [95]. This mechanism has also been observed for other targets, since KDM6B was involved in the stabilization of the transcription factor FOXO1 (Forkhead box protein O1, an inducer of pro apoptotic genes BIM, TRAIL, and FasL) in the nucleus of A549 and H460 non-small cell lung cancer cell lines. This interaction led to increased apoptosis and decreased migration and invasion [96] (Table 6).
3.4. New Anti-Cancer Therapeutic Protocols Targeting KDM6B Activity
Some pre-clinical studies already used the KDM6B inhibitors, GSKJ1/GSKJ4, to target cancers. GSKJ4 is a pro-drug for a cellular use of GSKJ1 when GSKJ1 is used in vitro This inhibitor has been identified by Kruidener and collaborators in 2012 [97]. In the acute lymphoblastic leukemia (ALL) cell line CUTLL1, GSKJ4 treatment induced cell cycle arrest and apoptosis and therefore is proposed as a promising therapeutic approach. However, this treatment did not affect myeloid leukemia cells, stromal cells, or hematopoietic progenitors [98]. GSKJ4 treatment also decreased tumor growth of xenografts of SF7761 and SF8628 pediatric brainstem glioma cells in mice. These cell lines present a heterozygous mutation of K27 of histone H3 (K27S) and half of histone H3 cannot be methylated at lysine 27 anymore. Nevertheless, mutated histone H3 sequestrated PRC2 and induced a dominant negative effect via reducing H3K27me3 on non-mutated histone H3. GSKJ4 treatment allowed the restoration of the mark H3K27me3 on histone H3, which was not mutated. GSKJ4 did not present the same effect on other pediatric brainstem cell lines, which did not contain the mutation [99].
4. Conclusions
This review highlights the paradoxical functions of EZH2 and KDM6B during EMT and cancer aggressiveness. Indeed, even if it has been shown that these proteins often present pro-EMT or pro-metastasis roles in a wide range of cancers, some examples revealed that they also presented anti-tumor functions in some cancers. Moreover, EZH2 and KDM6B were frequently both overexpressed and associated with EMT, invasion, migration, or metastasis in the same cancer types, such as ovarian cancers [36,82] or clear cell renal cell carcinomas [28,29,77]. These observations suggested that EZH2 and KDM6B expression might be regulated via shared similar mechanisms. However, to our knowledge, no studies targeted the common regulation of EZH2 and KDM6B during EMT and cancer. This review shows that the action of these two antagonistic enzymes during EMT is a complex phenomenon. The first paradoxical role of these enzymes is so that they are antagonistic, but both are involved in cancer aggressiveness and EMT sometimes in the same cell lines. The second paradoxical role is that these enzymes are also inactivated or under-expressed in some cancer types and still linked to epithelial phenotype in other cancer cell lines.
Therefore, all these studies suggested that EZH2 and KDM6B can be considered as master regulators of EMT through orchestrating the regulation of the H3K27me3 epigenetic mark. However, the mechanisms involved in this regulation remain unclear and seem often indirect and/or cell/cancer model-dependent. We think that the process called “type III EMT” gathers a lot of processes regulated by different pathways but lead to a common phenotype: the activation of EMT-ATFs expression and induction of a mesenchymal phenotype at both molecular and functional levels. We also know that cancer cells present a very variable genotype and that EZH2 and KDM6B could drive a specific cell phenotype depending on the cellular context. Moreover, interaction of EZH2 and KDM6B with different partners may also regulate their specific recruitment on promoters. Finally, H3K27me3 seems to be dynamically regulated during EMT and a slight change in the equilibrium between EZH2 and KDM6B expression, or activity, might rapidly modify the expression of the target genes. As described before, many studies have already focused on the regulation of EZH2 but the regulation of KDM6B remains largely unknown and may become a major issue to understand its function during EMT.
Therefore, the characterization of the role of EZH2 and KDM6B during EMT should provide new possibilities for designing anti-cancer therapeutic approaches. For example, in a recent study, Tazemetostat (an EZH2 chemical inhibitor which is used in many clinical trials) is more effective to target B-cell lymphoma cell lines models bearing EZH2 activating mutations than models bearing wild-type EZH2 since cell viability is less dependent on EZH2 in WT cells. Indeed, EZH2 inhibition led to the activation of B-cell pathway. A better understanding of the EZH2 target genes allowed the authors to associate Tazemetostat with B-cell pathway inhibitors to increase the specificity and the efficacy of this treatment in B-cell lymphoma with wild-type EZH2 [100].
Funding
Camille Lachat was supported by a fellowship from the MESR (Ministère de l’Enseignement Supérieur et de la Recherché). This work was supported by funding from institutional grants from INSERM (Institut National de la Santé et de la Recherche Médicale), EFS (Etablissement Français du Sang), and Univ. Bourgogne Franche-Comté and by the “Ligue Contre le Cancer” (001AC.2015) and the “Région Bourgogne Franche-Comté” (2014C-15449).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ADP | Adenosine Diphosphate |
| AKT | Protein Kinase B |
| ALL | Akute Lymphoblastic Leukemia |
| AMD1 | Adenosyl Methionine Decarboxylase 1 |
| APC | Adenomatous Polyposis Coli |
| ARF | Alternative Reading Frame |
| BIM | BCL2 Like 11 |
| BMP | Bone Morphogenetic Protein |
| CDH1 | Cadherin 1 |
| CDK | Cyclin Dependant Kinase |
| CEBPA | CCAAT/Enhancer Binding Protein α |
| ChIP | Chromatin ImmunoPrecipitation |
| DLBCL | Diffuse Large B Cell Lymphoma |
| DNA | Desoxyribonucleic Acid |
| EED | Embryonic Ectoderm Development |
| EGF | Epidermal Growth Factor |
| EMT | Epithelial to Mesenchymal Transition |
| EMT-ATF | Epithelial to Mesenchymal Transition Activating Transcription Factors |
| EPCAM | Epithelial Cell Adhesion Molecule |
| ER | Estrogen Receptor |
| ERK | Extracellular Signal-Regulated Kinase |
| EZH2 | Enhancer of Zeste Homolog 2 |
| EZH2i | Enhancer of Zeste Homolog 2 Inhibitor |
| FOS | FBJ Murine Osteosarcoma Viral Oncogene Homolog |
| FOXO1 | Forkhead Box Protein O1 |
| H2A | Histone 2A |
| H2B | Histone 2B |
| H3 | Histone 3 |
| H3K27Ac | Histone 3, Lysine 27 Acetylated |
| H3K27me2 | Histone 3, Lysine 27 Dimethylated |
| H3K27me3 | Histone 3, Lysine 27 Trimethylated |
| H3K27me3 | Histone 3, Lysine 27 Methylated |
| HCC | Hepatocellular Carcinoma |
| HDAC1 | Histone Deacetylase 1 |
| HDAC2 | Histone Deacetylase 2 |
| HER2 | Human Epidermal Growth Factor Receptor |
| HOTAIR | HOX Transcript Antisense RNA |
| HOX | Homeobox |
| IF | ImmunoFluorescence |
| IGF1 | Insulin-Like Growth Factor 1 |
| IHC | Immunohistochemistry |
| INK4 | Inhibitor of CDK4 |
| JMJD3 | Jumonji Domain-Containing Protein 3 |
| JUB1 | JUNGBRUNNEN 1 |
| KDM6A | Lysine Demethylase 6A |
| KDM6B | Lysine Demethylase 6B |
| KMT6A | Lysine Methyltransferase 6A |
| KO | Knock-Out |
| KRAS | V-Ki-Ras2 Kirsten Rat Sarcoma Viral Oncogene Homolog |
| lncRNA | Long Non-Coding RiboNucleic Acid |
| MAPK | Mitogen-Activated Protein Kinase |
| MEK | Mitogen-Activated Protein Kinase |
| MET | Mesenchymal to Epithelial Transition |
| miRNA | Micro Ribonucleic Acid |
| MM | Multiple Myeloma |
| MPM | Multiple Pleural Mesothelioma |
| mRNA | Messenger RiboNucleic Acid |
| N-CAD | N-Cadherin |
| NF-κB | Nuclear Factor-κB |
| NHL | Non-Hodgkin Lymphoma |
| OCT4 | Octamer-Binding Transcription Factor 4 |
| PDAC | Pancreatic Ductile Adenocarcinoma |
| PD-L1 | Programmed Death Ligand 1 |
| PRC2 | Polycomb Repressive Complex 2 |
| PRE | Polycomb Repressive Complex 2 Response Element |
| RBBP4 | RetinoBlastoma Binding Protein 4 |
| RBBP7 | RetinoBlastoma Binding Protein 7 |
| RelA | V-Rel Avian Reticuloendotheliosis Viral Oncogene Homolog A |
| RelB | V-Rel Avian Reticuloendotheliosis Viral Oncogene Homolog B |
| RKIP | Raf-1 Kinas Inhibitor Protein |
| RNA | Ribonucleic Acid |
| RT-PCR | Reverse Transcription Polymerase Chain Reaction |
| RT-qPCR | Reverse Transcription Quantitative Polymerase Chain Reaction |
| siRNA | Small Interfering RiboNucleic Acid |
| SOX4 | SRY-Related HMG Box |
| SUZ-12 | Suppressor of Zeste 12 |
| TF | Transcription Factor |
| tFL | Transformed Follicular Lymphoma |
| TGF-β | Transforming Growth Factor β |
| TIC | Tumor Initiating Cells |
| TNF-α | Tumor Necrosis Factor α |
| TRAIL | Tumor Necrosis Factor Related Apoptosis-Inducing Ligand |
| UTR | Untranslated Region |
| VIM | Vimentin |
| ZEB | Zinc Finger E-box Binding Homeobox |
References
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; André, F.; de Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Janji, B.; Tittarelli, A.; Eggermont, A.; Thiery, J.P. Tumor plasticity interferes with anti-tumor immunity. Crit. Rev. Immunol. 2014, 34, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Price, L.S.; Collard, J.G. Regulation of the cytoskeleton by Rho-family GTPases: Implications for tumour cell invasion. Semin. Cancer Biol. 2001, 11, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Betson, M.; Braga, V.M.M. Tumor progression: Small GTPases and loss of cell–cell adhesion. BioEssays 2003, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Papiewska-Pająk, I.; Kowalska, M.A.; Boncela, J. Expression and activity of SNAIL transcription factor during Epithelial to Mesenchymal Transition (EMT) in cancer progression. Postepy Hig. Med. Doswiadczalnej Online 2016, 70, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, P.; Bissell, M.J.; Radisky, D.C. Epithelial-Mesenchymal Transition: General Principles and Pathological Relevance with Special Emphasis on the Role of Matrix Metalloproteinases. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef]
- Sing, A.; Pannell, D.; Karaiskakis, A.; Sturgeon, K.; Djabali, M.; Ellis, J.; Lipshitz, H.D.; Cordes, S.P. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell 2009, 138, 885–897. [Google Scholar] [CrossRef]
- Ye, L.; Fan, Z.; Yu, B.; Chang, J.; Hezaimi, K.A.; Zhou, X.; Park, N.-H.; Wang, C.-Y. Histone Demethylases KDM4B and KDM6B Promotes Osteogenic Differentiation of Human MSCs. Cell Stem Cell 2012, 11, 50–61. [Google Scholar] [CrossRef]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Rappsilber, J.; Nielsen, A.L.; Johansen, J.V.; Helin, K. The Histone Lysine Demethylase JMJD3/KDM6B Is Recruited to p53 Bound Promoters and Enhancer Elements in a p53 Dependent Manner. PLoS ONE 2014, 9, e96545. [Google Scholar] [CrossRef] [PubMed]
- Burchfield, J.S.; Li, Q.; Wang, H.Y.; Wang, R.-F. JMJD3 as an epigenetic regulator in development and disease. Int. J. Biochem. Cell Biol. 2015, 67, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yan, Y.; Wang, X.; Jiang, Y.; Xu, H.E. EZH2: Biology, disease, and structure-based drug discovery. Acta Pharmacol. Sin. 2014, 35, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Perrigue, P.M.; Najbauer, J.; Barciszewski, J. Histone demethylase JMJD3 at the intersection of cellular senescence and cancer. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Daures, M.; Ngollo, M.; Judes, G.; Rifaï, K.; Kemeny, J.-L.; Penault-Llorca, F.; Bignon, Y.-J.; Guy, L.; Bernard-Gallon, D. The JMJD3 Histone Demethylase and the EZH2 Histone Methyltransferase in Prostate Cancer. Omics J. Integr. Biol. 2016, 20, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Daures, M.; Idrissou, M.; Judes, G.; Rifaï, K.; Penault-Llorca, F.; Bignon, Y.-J.; Guy, L.; Bernard-Gallon, D. A new metabolic gene signature in prostate cancer regulated by JMJD3 and EZH2. Oncotarget 2018, 9, 23413–23425. [Google Scholar] [CrossRef]
- Lee, H.W.; Choe, M. Expression of EZH2 in renal cell carcinoma as a novel prognostic marker. Pathol. Int. 2012, 62, 735–741. [Google Scholar] [CrossRef]
- Xu, B.; Abourbih, S.; Sircar, K.; Kassouf, W.; Mansure, J.J.; Aprikian, A.; Tanguay, S.; Brimo, F. Enhancer of zeste homolog 2 expression is associated with metastasis and adverse clinical outcome in clear cell renal cell carcinoma: a comparative study and review of the literature. Arch. Pathol. Lab. Med. 2013, 137, 1326–1336. [Google Scholar] [CrossRef]
- Yamada, A.; Fujii, S.; Daiko, H.; Nishimura, M.; Chiba, T.; Ochiai, A. Aberrant expression of EZH2 is associated with a poor outcome and P53 alteration in squamous cell carcinoma of the esophagus. Int. J. Oncol. 2011, 38, 345–353. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 Expression Is Associated With High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.; Solis, L.M.; Lin, H.; Yuan, P.; Tang, X.; Kadara, H.; Riquelme, E.; Galindo, H.; Moran, C.A.; Kalhor, N.; et al. EZH2 Protein Expression Associates With the Early Pathogenesis, Tumor Progression and Prognosis of Non-small Cell Lung Carcinoma. Clin. Cancer Res. 2013, 19, 6556–6565. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Hurt, E.M.; Farrar, W.L. Clinical significance of Polycomb gene expression in brain tumors. Mol. Cancer 2010, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, Y.; Semba, S.; Kato, H.; Ito, A.; Yanagihara, K.; Yokozaki, H. Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 2006, 97, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.-Y.; Cai, M.-Y.; Yang, G.-F.; He, L.-R.; Mai, S.-J.; Hua, W.-F.; Liao, Y.-J.; Deng, H.-X.; Chen, Y.-C.; Guan, X.-Y.; et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Albadine, R.; Magheli, A.; Guzzo, T.J.; Ball, M.W.; Hinz, S.; Schoenberg, M.P.; Netto, G.J.; Gonzalgo, M.L. Increased EZH2 protein expression is associated with invasive urothelial carcinoma of the bladder. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Weikert, S.; Christoph, F.; Köllermann, J.; Müller, M.; Schrader, M.; Miller, K.; Krause, H. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int. J. Mol. Med. 2005, 16, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hou, N.; Cheng, X.; Zhang, J.; Tan, X.; Zhang, C.; Tang, Y.; Teng, Y.; Yang, X. Ezh2 Acts as a Tumor Suppressor in Kras-driven Lung Adenocarcinoma. Int. J. Biol. Sci. 2017, 13, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Muto, T.; Sashida, G.; Oshima, M.; Wendt, G.R.; Mochizuki-Kashio, M.; Nagata, Y.; Sanada, M.; Miyagi, S.; Saraya, A.; Kamio, A.; et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 2013, 210, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Gwak, S.Y.; Shim, G.-A.; Liu, L.; Lim, Y.C.; Kim, J.M.; Jung, M.G.; Koo, B.S. EZH2 is associated with poor prognosis in head-and-neck squamous cell carcinoma via regulating the epithelial-to-mesenchymal transition and chemosensitivity. Oral Oncol. 2016, 52, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Zhao, J.; Vieth, E.; Nephew, K.P.; Matei, D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget 2016, 7, 84453–84467. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef]
- Xia, H.; Zhang, W.; Zhang, B.; Zhao, Y.; Zhao, Y.; Li, S.; Liu, Y. miR-21 modulates the effect of EZH2 on the biological behavior of human lung cancer stem cells in vitro. Oncotarget 2017, 8, 85442–85451. [Google Scholar] [CrossRef]
- Cakouros, D.; Isenmann, S.; Cooper, L.; Zannettino, A.; Anderson, P.; Glackin, C.; Gronthos, S. Twist-1 Induces Ezh2 Recruitment Regulating Histone Methylation along the Ink4A/Arf Locus in Mesenchymal Stem Cells. Mol. Cell. Biol. 2012, 32, 1433–1441. [Google Scholar] [CrossRef]
- Zheng, M.; Jiang, Y.; Chen, W.; Li, K.; Liu, X.; Gao, S.; Feng, H.; Wang, S.; Jiang, J.; Ma, X.; et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget 2015, 6, 6797–6810. [Google Scholar] [CrossRef]
- Tong, Z.-T.; Cai, M.-Y.; Wang, X.-G.; Kong, L.-L.; Mai, S.-J.; Liu, Y.-H.; Zhang, H.-B.; Liao, Y.-J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef]
- Herranz, N.; Pasini, D.; Díaz, V.M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb Complex 2 Is Required for E-cadherin Repression by the Snail1 Transcription Factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef]
- CHO, H.M.; JEON, H.S.; LEE, S.Y.; JEONG, K.J.; PARK, S.-Y.; LEE, H.Y.; LEE, J.U.; KIM, J.H.; KWON, S.J.; CHOI, E.; et al. microRNA-101 inhibits lung cancer invasion through the regulation of enhancer of zeste homolog 2. Exp. Ther. Med. 2011, 2, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Zhu, H.; Song, H.; Wang, Z.; Huang, G.; Li, D.; Ma, Z.; Ma, J.; Qin, Q.; Sun, X.; et al. Targets and molecular mechanisms of triptolide in cancer therapy. Chin. J. Cancer Res. 2014, 26, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Tamgue, O.; Chai, C.-S.; Hao, L.; Zambe, J.-C.D.; Huang, W.-W.; Zhang, B.; Lei, M.; Wei, Y.-M. Triptolide inhibits histone methyltransferase EZH2 and modulates the expression of its target genes in prostate cancer cells. Asian Pac. J. Cancer Prev. APJCP 2013, 14, 5663–5669. [Google Scholar] [CrossRef] [PubMed]
- Martinho, O.; Pinto, F.; Granja, S.; Miranda-Gonçalves, V.; Moreira, M.A.R.; Ribeiro, L.F.J.; di Loreto, C.; Rosner, M.R.; Longatto-Filho, A.; Reis, R.M. RKIP Inhibition in Cervical Cancer Is Associated with Higher Tumor Aggressive Behavior and Resistance to Cisplatin Therapy. PLoS ONE 2013, 8, e59104. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, S.; Duan, M.; Moen, E.L.; Cross-Knorr, S.; Brilliant, K.; Bonavida, B.; LaValle, T.; Yeung, K.C.; Al-Mulla, F.; Chin, E.; et al. Raf Kinase Inhibitor Protein (RKIP) Blocks Signal Transducer and Activator of Transcription 3 (STAT3) Activation in Breast and Prostate Cancer. PLoS ONE 2014, 9, e92478. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, P.; Li, B.; Sun, P.; Zhang, J.; Wang, B.; Jia, B. RKIP suppresses gastric cancer cell proliferation and invasion and enhances apoptosis regulated by microRNA-224. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 10095–10103. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Baritaki, S.; Marathe, H.; Feng, J.; Park, S.; Beach, S.; Bazeley, P.S.; Beshir, A.B.; Fenteany, G.; Mehra, R.; et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 2012, 72, 3091–3104. [Google Scholar] [CrossRef]
- Sato, T.; Kaneda, A.; Tsuji, S.; Isagawa, T.; Yamamoto, S.; Fujita, T.; Yamanaka, R.; Tanaka, Y.; Nukiwa, T.; Marquez, V.E.; et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of Pancreatic Tumor Cell Proliferation and Chemoresistance by the Histone Methyltransferase EZH2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef]
- Fu, X.; Lin, J.; Qin, F.; Yang, Z.; Ding, Y.; Zhang, Y.; Han, L.; Zhu, X.; Zhang, Q. LncAPC drives Wnt/β-catenin activation and liver TIC self-renewal through EZH2 mediated APC transcriptional inhibition. Mol. Carcinog. 2017. [Google Scholar] [CrossRef]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.M.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef]
- Shi, B.; Liang, J.; Yang, X.; Wang, Y.; Zhao, Y.; Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Li, R.; et al. Integration of Estrogen and Wnt Signaling Circuits by the Polycomb Group Protein EZH2 in Breast Cancer Cells. Mol. Cell. Biol. 2007, 27, 5105–5119. [Google Scholar] [CrossRef]
- Tiwari, N.; Tiwari, V.K.; Waldmeier, L.; Balwierz, P.J.; Arnold, P.; Pachkov, M.; Meyer-Schaller, N.; Schübeler, D.; van Nimwegen, E.; Christofori, G. Sox4 Is a Master Regulator of Epithelial-Mesenchymal Transition by Controlling Ezh2 Expression and Epigenetic Reprogramming. Cancer Cell 2013, 23, 768–783. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Tokita, K.; Wada, N.; Ito, K.; Yamauchi, C.; Ito, Y.; Ochiai, A. MEK–ERK pathway regulates EZH2 overexpression in association with aggressive breast cancer subtypes. Oncogene 2011, 30, 4118–4128. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Mani, R.-S.; Russo, N.; Scanlon, C.S.; Tsodikov, A.; Jing, X.; Cao, Q.; Palanisamy, N.; Metwally, T.; Inglehart, R.C.; et al. The tumor suppressor gene rap1GAP is silenced by mir-101-mediated EZH2 overexpression in invasive squamous cell carcinoma. Oncogene 2011, 30, 4339–4349. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.; Nilsson, J.; Mir, S.E.; van der Stoop, P.M.; Hulleman, E.; Niers, J.M.; de Witt Hamer, P.C.; Marquez, V.E.; Cloos, J.; Krichevsky, A.M.; et al. miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget 2011, 1, 710–720. [Google Scholar]
- Wang, H.-J.; Ruan, H.-J.; He, X.-J.; Ma, Y.-Y.; Jiang, X.-T.; Xia, Y.-J.; Ye, Z.-Y.; Tao, H.-Q. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur. J. Cancer 2010, 46, 2295–2303. [Google Scholar] [CrossRef]
- Cui, S.; Sun, Y.; Liu, Y.; Liu, C.; Wang, J.; Hao, G.; Sun, Q. MicroRNA-137 has a suppressive role in liver cancer via targeting EZH2. Mol. Med. Rep. 2017, 16, 9494–9502. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.; Chen, Z.; Jin, Y.; Wang, Y.; Kolokythas, A.; Dai, Y.; Zhou, X. MicroRNA-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem. J. 2011, 440, 23–31. [Google Scholar] [CrossRef]
- Zheng, F.; Liao, Y.-J.; Cai, M.-Y.; Liu, Y.-H.; Liu, T.-H.; Chen, S.-P.; Bian, X.-W.; Guan, X.-Y.; Lin, M.C.; Zeng, Y.-X.; et al. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut 2012, 61, 278–289. [Google Scholar] [CrossRef]
- Huang, S.-D.; Yuan, Y.; Zhuang, C.-W.; Li, B.-L.; Gong, D.-J.; Wang, S.-G.; Zeng, Z.-Y.; Cheng, H.-Z. MicroRNA-98 and microRNA-214 post-transcriptionally regulate enhancer of zeste homolog 2 and inhibit migration and invasion in human esophageal squamous cell carcinoma. Mol. Cancer 2012, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ooi, L.L.P.J.; Hui, K.M. MiR-214 Targets β-Catenin Pathway to Suppress Invasion, Stem-Like Traits and Recurrence of Human Hepatocellular Carcinoma. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Sun, J.; Tian, X.; Lu, S.-Q.; Hu, H.-B. MicroRNA-4465 suppresses tumor proliferation and metastasis in non-small cell lung cancer by directly targeting the oncogene EZH2. Biomed. Pharmacother. Biomedecine Pharmacother. 2017. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Tetteh, P.W.; Merz, P.R.; Dickes, E.; Abukiwan, A.; Hotz-Wagenblatt, A.; Holland-Cunz, S.; Sinnberg, T.; Schittek, B.; Schadendorf, D.; et al. miR-137 Inhibits the Invasion of Melanoma Cells through Downregulation of Multiple Oncogenic Target Genes. J. Invest. Dermatol. 2013, 133, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Winter, J.N.; Leonard, J.P.; Ribrag, V.; Constantinidou, A.; Giulino-Roth, L.; Michot, J.-M.; Khan, T.A.; Horner, T.; Carver, J.; et al. A Phase I Study of GSK2816126, an Enhancer of Zeste Homolog 2(EZH2) Inhibitor, in Patients (pts) with Relapsed/Refractory Diffuse Large B-Cell Lymphoma (DLBCL), Other Non-Hodgkin Lymphomas (NHL), Transformed Follicular Lymphoma (tFL), Solid Tumors and Multiple Myeloma (MM). Blood 2016, 128, 4203. [Google Scholar]
- Tang, B.; Qi, G.; Tang, F.; Yuan, S.; Wang, Z.; Liang, X.; Li, B.; Yu, S.; Liu, J.; Huang, Q.; et al. Aberrant JMJD3 Expression Upregulates Slug to Promote Migration, Invasion, and Stem Cell–Like Behaviors in Hepatocellular Carcinoma. Cancer Res. 2016, 76, 6520–6532. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hou, L.; Ding, G.; Li, Y.; Wang, J.; Qian, B.; Sun, J.; Wang, Q. KDM6B induces epithelial-mesenchymal transition and enhances clear cell renal cell carcinoma metastasis through the activation of SLUG. Int. J. Clin. Exp. Pathol. 2015, 8, 6334–6344. [Google Scholar]
- Ohguchi, H.; Harada, T.; Sagawa, M.; Kikuchi, S.; Tai, Y.-T.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 2017. [Google Scholar] [CrossRef]
- Anderton, J.A.; Bose, S.; Vockerodt, M.; Vrzalikova, K.; Wei, W.; Kuo, M.; Helin, K.; Christensen, J.; Rowe, M.; Murray, P.G.; et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein–Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 2011, 30, 2037–2043. [Google Scholar] [CrossRef]
- Ramadoss, S.; Chen, X.; Wang, C.-Y. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 44508–44517. [Google Scholar] [CrossRef]
- Cregan, S.; Breslin, M.; Roche, G.; Wennstedt, S.; MacDonagh, L.; Albadri, C.; Gao, Y.; O’Byrne, K.J.; Cuffe, S.; Finn, S.P.; et al. Kdm6a and Kdm6b: Altered expression in malignant pleural mesothelioma. Int. J. Oncol. 2017, 50, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Yao, Q.; Wei, D.; Liu, M.; Geng, F.; Wang, Q.; Wang, Y. KDM6B promotes ovarian cancer cell migration and invasion by induced transforming growth factor-β1 expression. J. Cell. Biochem. 2019, 120, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Sato, T.; Yamamoto, S.; Miyabayashi, K.; Matsusaka, K.; Asaoka, Y.; Ijichi, H.; Hirata, Y.; et al. Loss of histone demethylase KDM6B enhances aggressiveness of pancreatic cancer through downregulation of C/EBPα. Carcinogenesis 2014, 35, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Barbáchano, A.; Silva, J.; Bonilla, F.; Campbell, M.J.; Muñoz, A.; Larriba, M.J. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 2011, 20, 4655–4665. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, R.; Sakamoto, Y.; Nakagawa, S.; Miyake, K.; Izumi, D.; Kosumi, K.; Taki, K.; Higashi, T.; Imamura, Y.; Ishimoto, T.; et al. The Prognostic Significance of Histone Lysine Demethylase JMJD3/KDM6B in Colorectal Cancer. Ann. Surg. Oncol. 2016, 23, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Yao, L.; Zhang, Q.; Wang, F.; Mei, H.; Guo, X.; Huang, W. Long noncoding RNA HOTAIR promotes metastasis of renal cell carcinoma by up-regulating histone H3K27 demethylase JMJD3. Oncotarget 2017, 8, 19795–19802. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.-Y.; Kuo, M.Y.-P.; Lu, T.-Y.; Wang, Y.-P.; Wu, H.-C. Generation of an anti-EpCAM antibody and epigenetic regulation of EpCAM in colorectal cancer. Int. J. Oncol. 2015, 46, 1788–1800. [Google Scholar] [CrossRef]
- Zhou, F.Q.; Qi, Y.M.; Xu, H.; Wang, Q.Y.; Gao, X.S.; Guo, H.G. Expression of EpCAM and Wnt/ β-catenin in human colon cancer. Genet. Mol. Res. GMR 2015, 14, 4485–4494. [Google Scholar] [CrossRef]
- Mo, J.; Wang, L.; Huang, X.; Lu, B.; Zou, C.; Wei, L.; Chu, J.; Eggers, P.K.; Chen, S.; Raston, C.L.; et al. Multifunctional nanoparticles for co-delivery of paclitaxel and carboplatin against ovarian cancer by inactivating the JMJD3-HER2 axis. Nanoscale 2017, 9, 13142–13152. [Google Scholar] [CrossRef]
- Yan, N.; Xu, L.; Wu, X.; Zhang, L.; Fei, X.; Cao, Y.; Zhang, F. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp. Cell Res. 2017, 359, 405–414. [Google Scholar] [CrossRef]
- Park, W.-Y.; Hong, B.-J.; Lee, J.; Choi, C.; Kim, M.-Y. H3K27 Demethylase JMJD3 Employs the NF-κB and BMP Signaling Pathways to Modulate the Tumor Microenvironment and Promote Melanoma Progression and Metastasis. Cancer Res. 2016, 76, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.-M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Ng, H.L.H.; Pan, W.; Chen, H.; Zhang, G.; Bian, Z.; Lu, A.; Yang, Z. Exploring Different Strategies for Efficient Delivery of Colorectal Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26936–26952. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.C.; Rudkjær, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Ene, C.I.; Edwards, L.; Riddick, G.; Baysan, M.; Woolard, K.; Kotliarova, S.; Lai, C.; Belova, G.; Cam, M.; Walling, J.; et al. Histone Demethylase Jumonji D3 (JMJD3) as a Tumor Suppressor by Regulating p53 Protein Nuclear Stabilization. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, N.; Zhang, Y.; Wang, C.; Ge, T.; Jin, H.; Deng, X.; Huo, X.; Gu, D.; Ge, Z.; et al. KDM6B Elicits Cell Apoptosis by Promoting Nuclear Translocation of FOXO1 in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2015, 37, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Chung, C.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Brach, D.; Johnston-Blackwell, D.; Drew, A.; Lingaraj, T.; Motwani, V.; Warholic, N.M.; Feldman, I.; Plescia, C.; Smith, J.J.; Copeland, R.A.; et al. EZH2 Inhibition by Tazemetostat Results in Altered Dependency on B-cell Activation Signaling in DLBCL. Mol. Cancer Ther. 2017, 16, 2586–2597. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).