
Integrated Multi-Omics Analysis of the Developmental Stages of Antheraea pernyi Pupae: Dynamic Changes in Metabolite Profiles and Gene Expression


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Metabolomics Analysis
2.2.1. Metabolite Extraction
2.2.2. Chromatographic Separation
2.2.3. Mass Spectrometry Acquisition
2.2.4. Data Preprocessing
2.3. Transcriptomics Analysis
2.3.1. RNA Extraction
2.3.2. Construction of RNA-Seq Libraries
2.3.3. Bioinformatics Analysis
3. Results
3.1. Metabolomics Analysis
3.1.1. Overall Metabolomics Analysis of Samples at Different Growth Stages
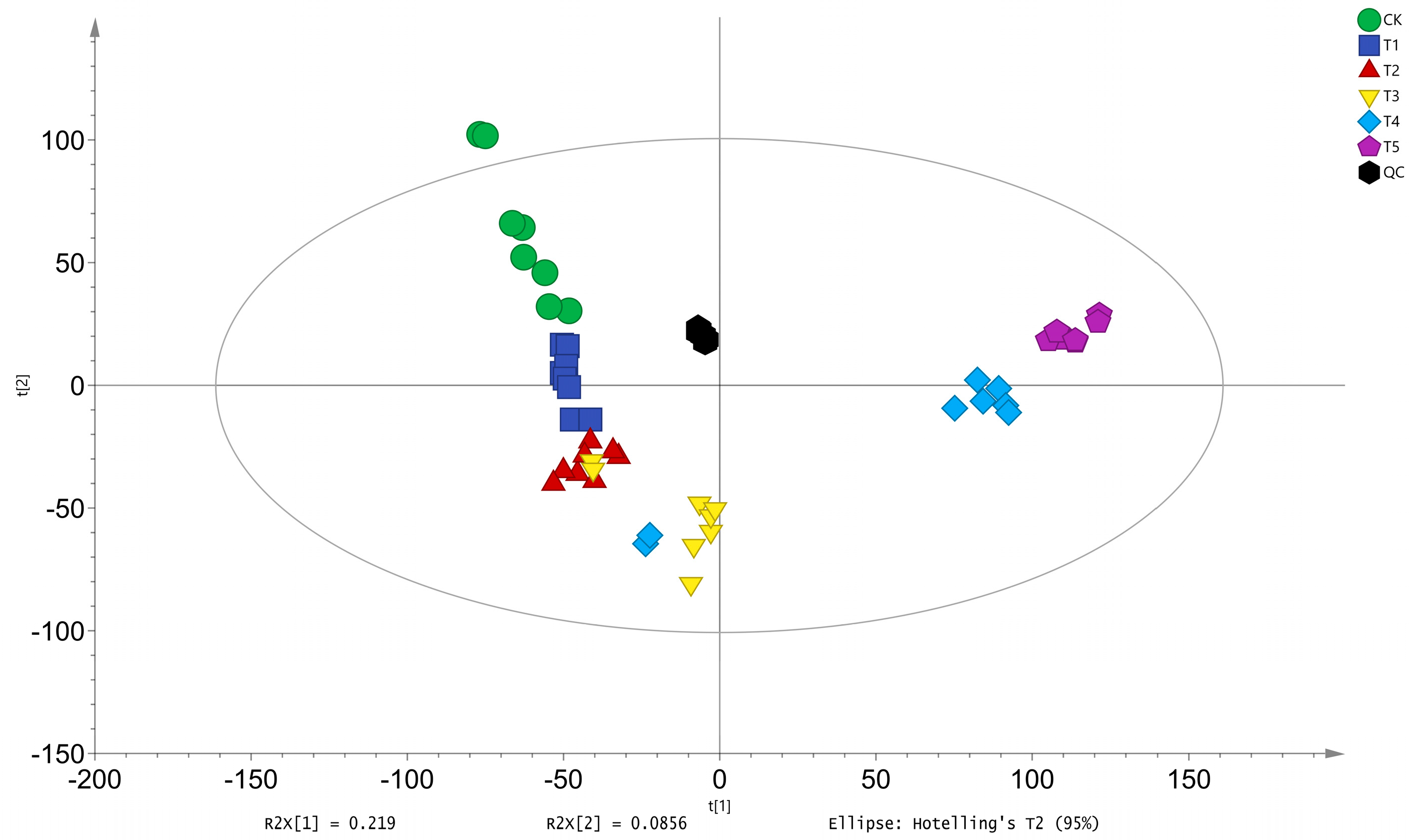
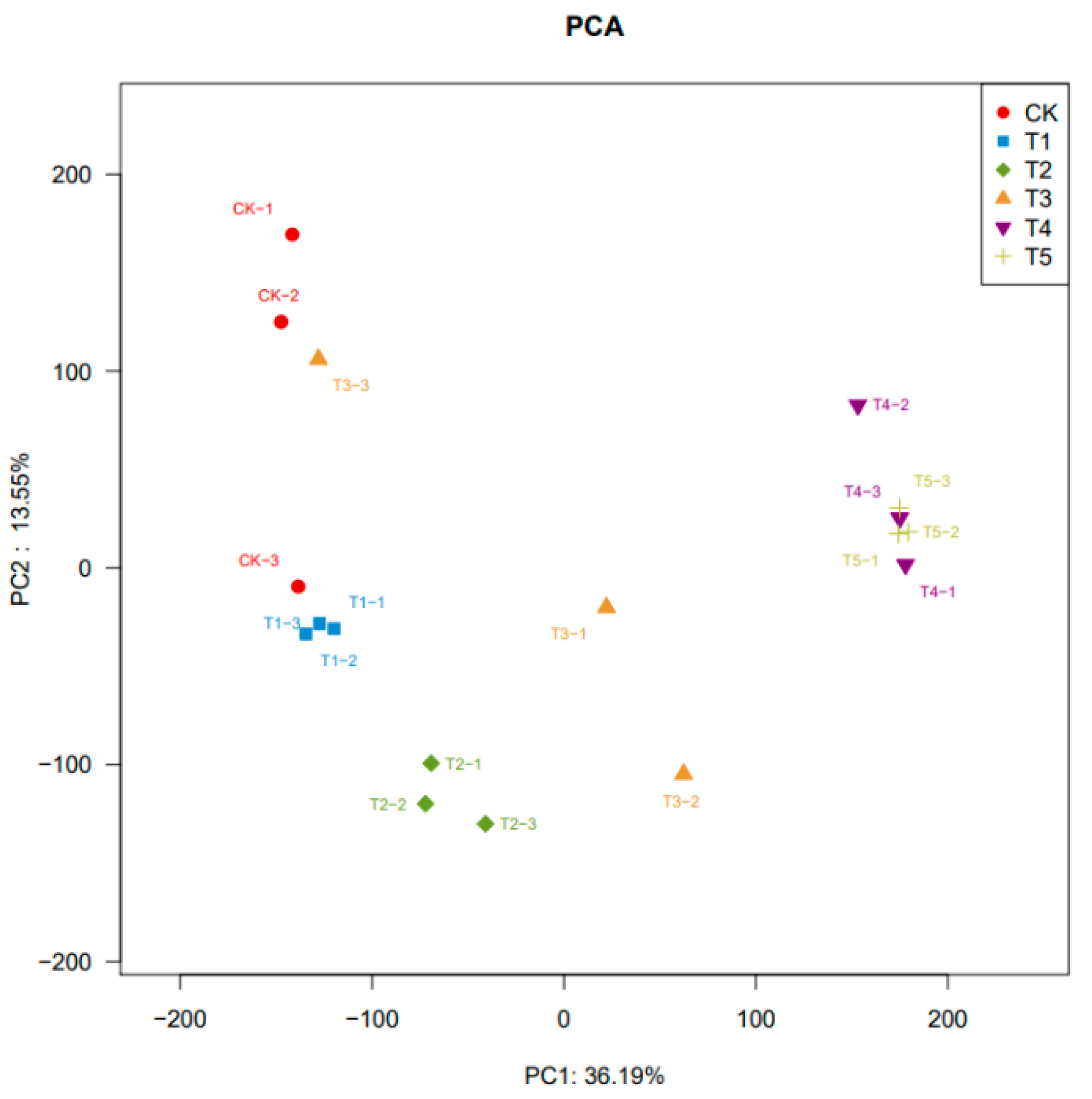
3.1.2. Principal Component Analysis
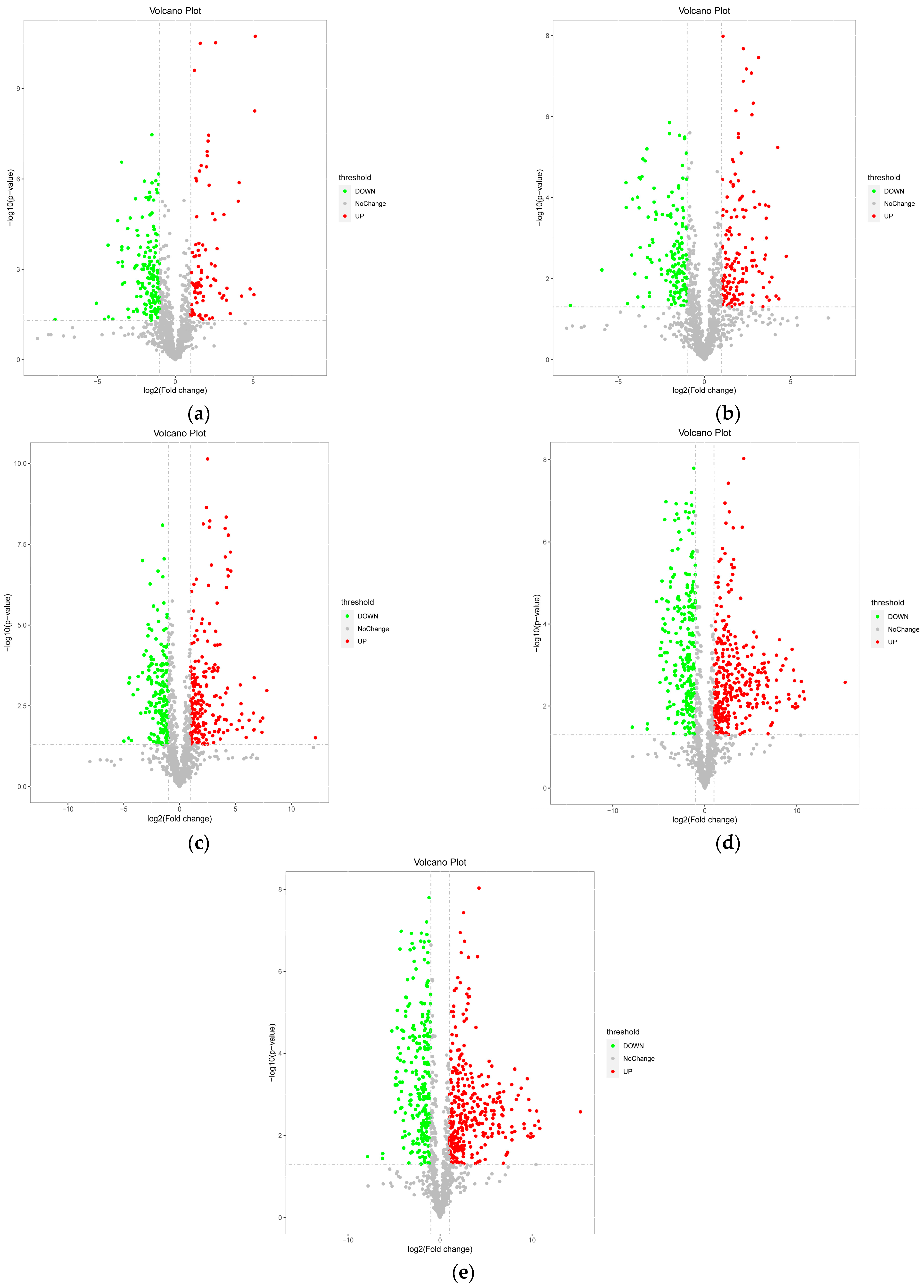
3.1.3. Univariate Statistical Analysis of Differential Metabolites at Different Developmental Stages
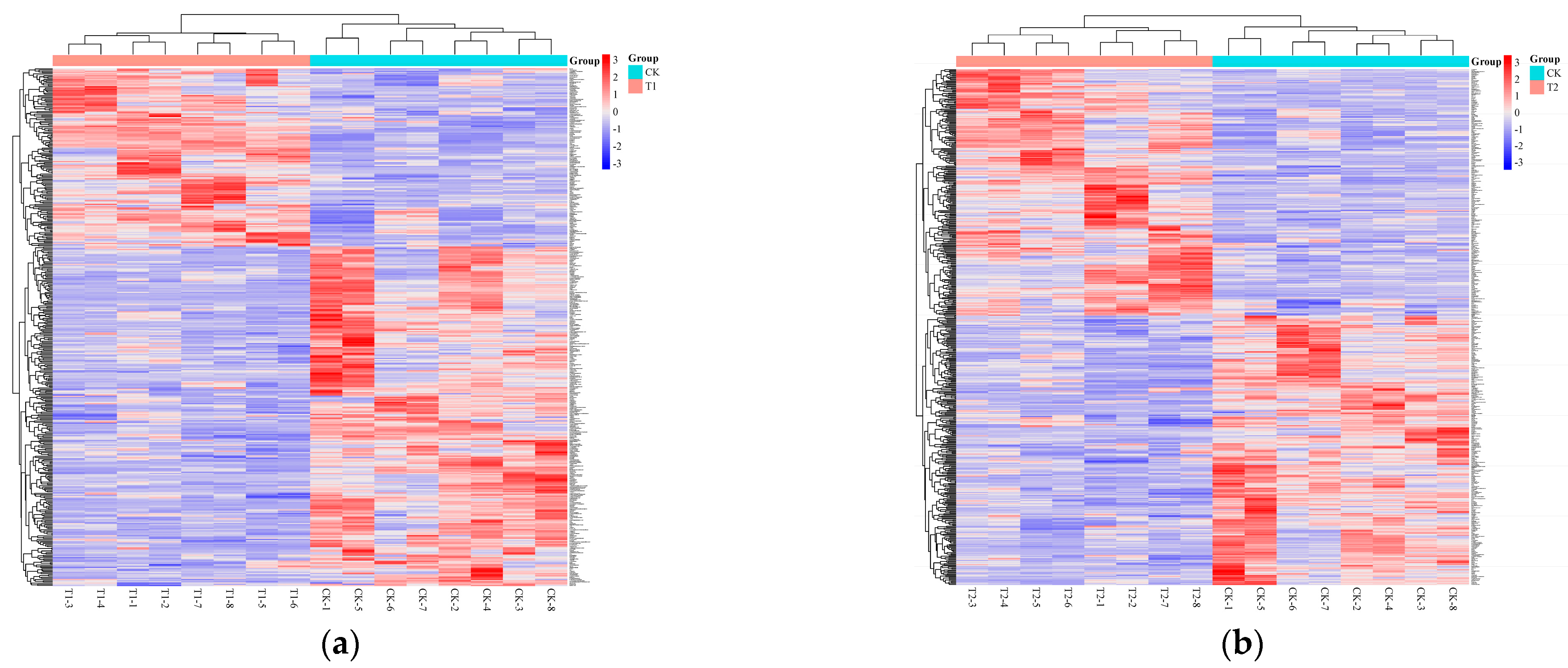
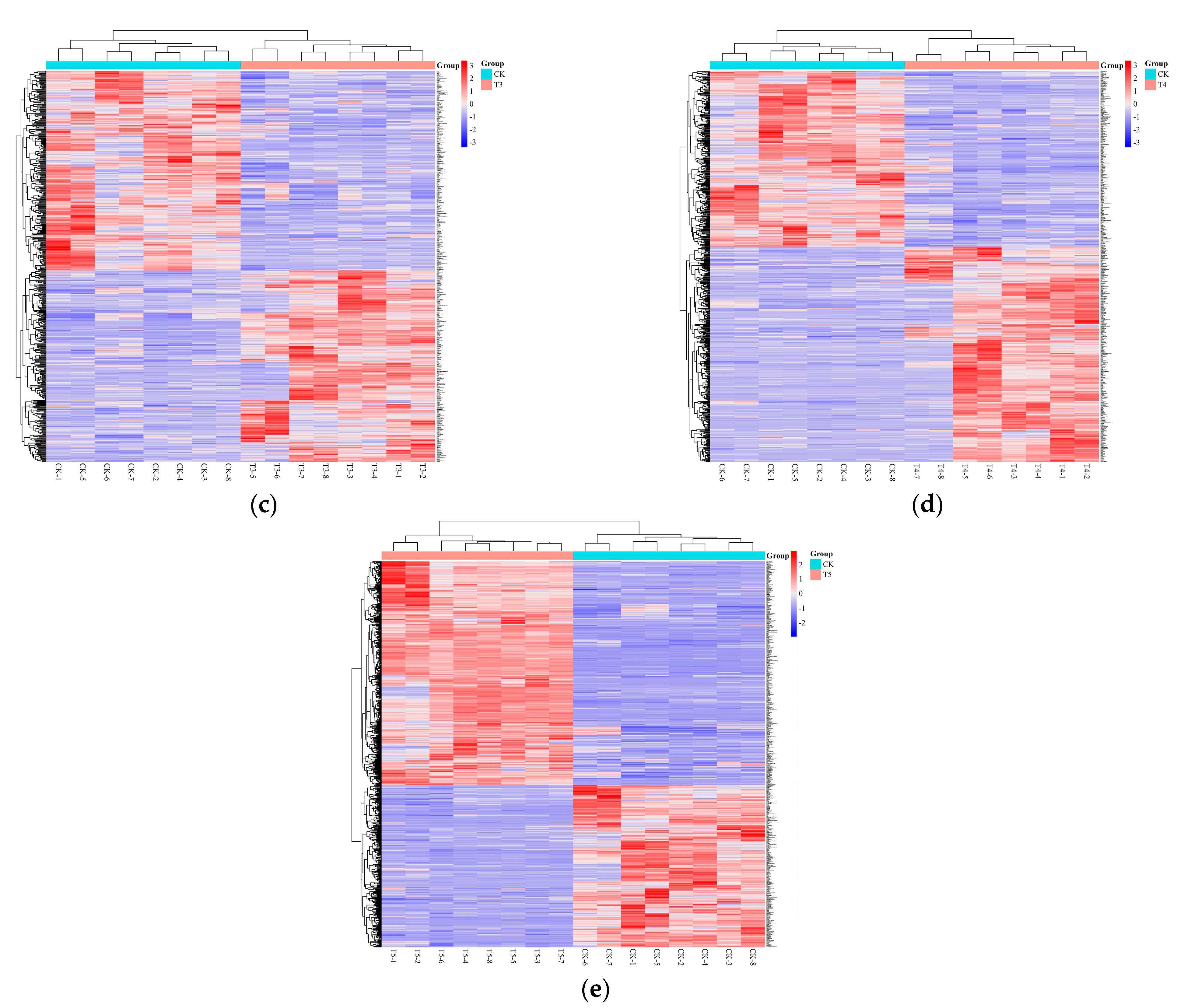
3.1.4. Hierarchical Clustering Analysis
3.1.5. Significant Differential Metabolites at Different Developmental Stages
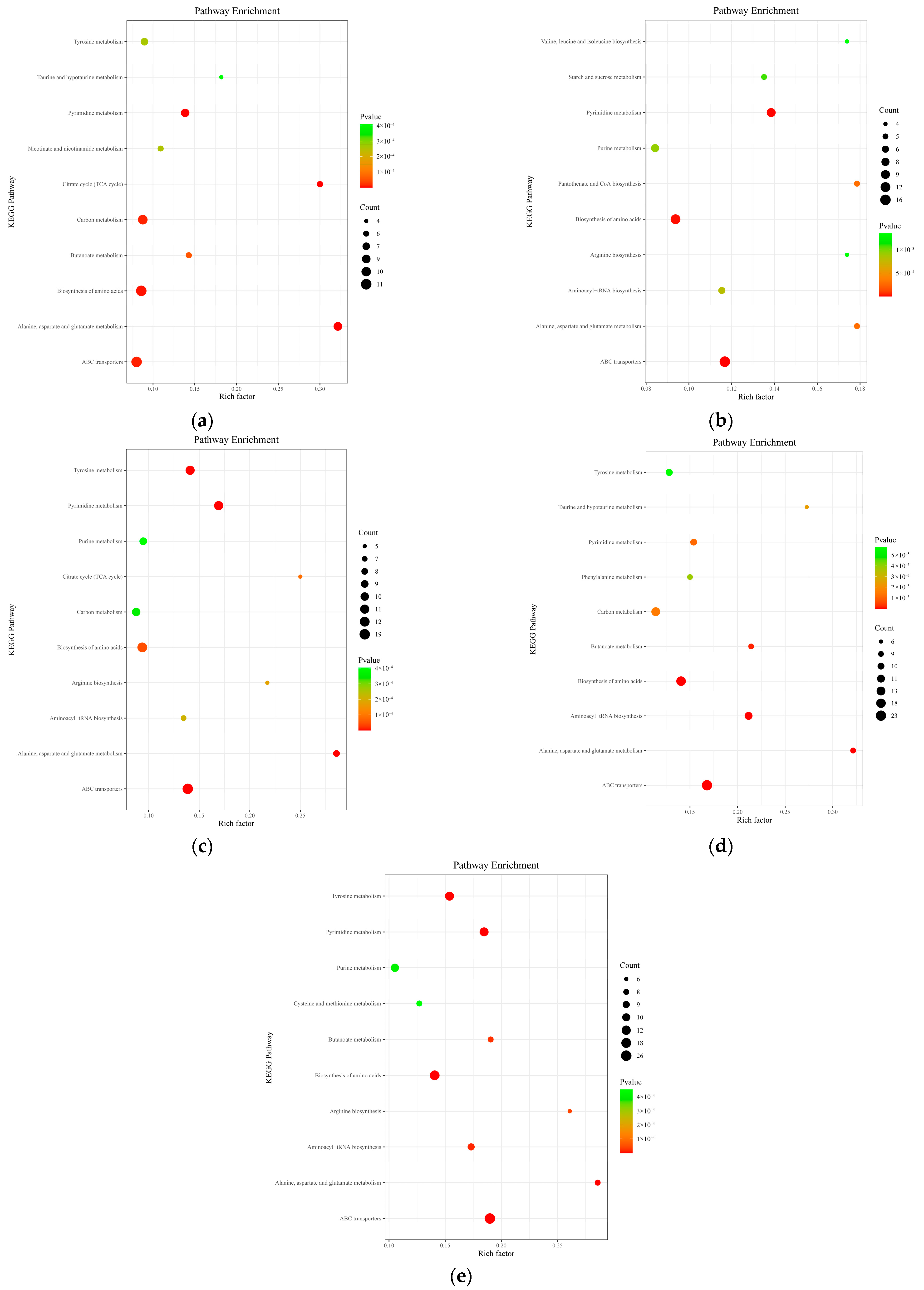
3.1.6. KEGG Metabolic Pathway Analysis
3.2. Transcriptomics Analysis
3.2.1. Analysis of Differentially Expressed Genes in Different Developmental Stages of A. pernyi Pupae
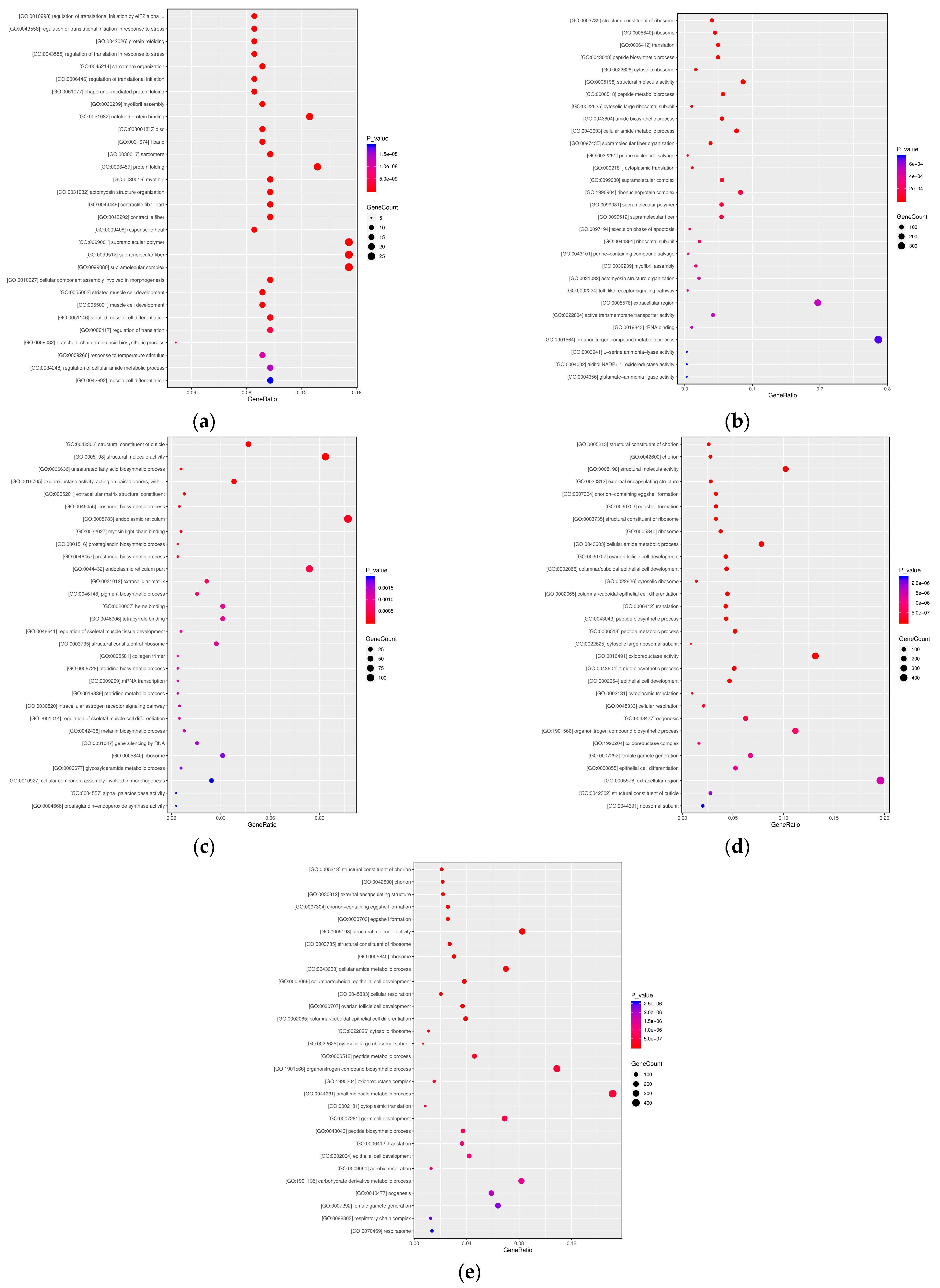
3.2.2. GO Functional Enrichment Analysis
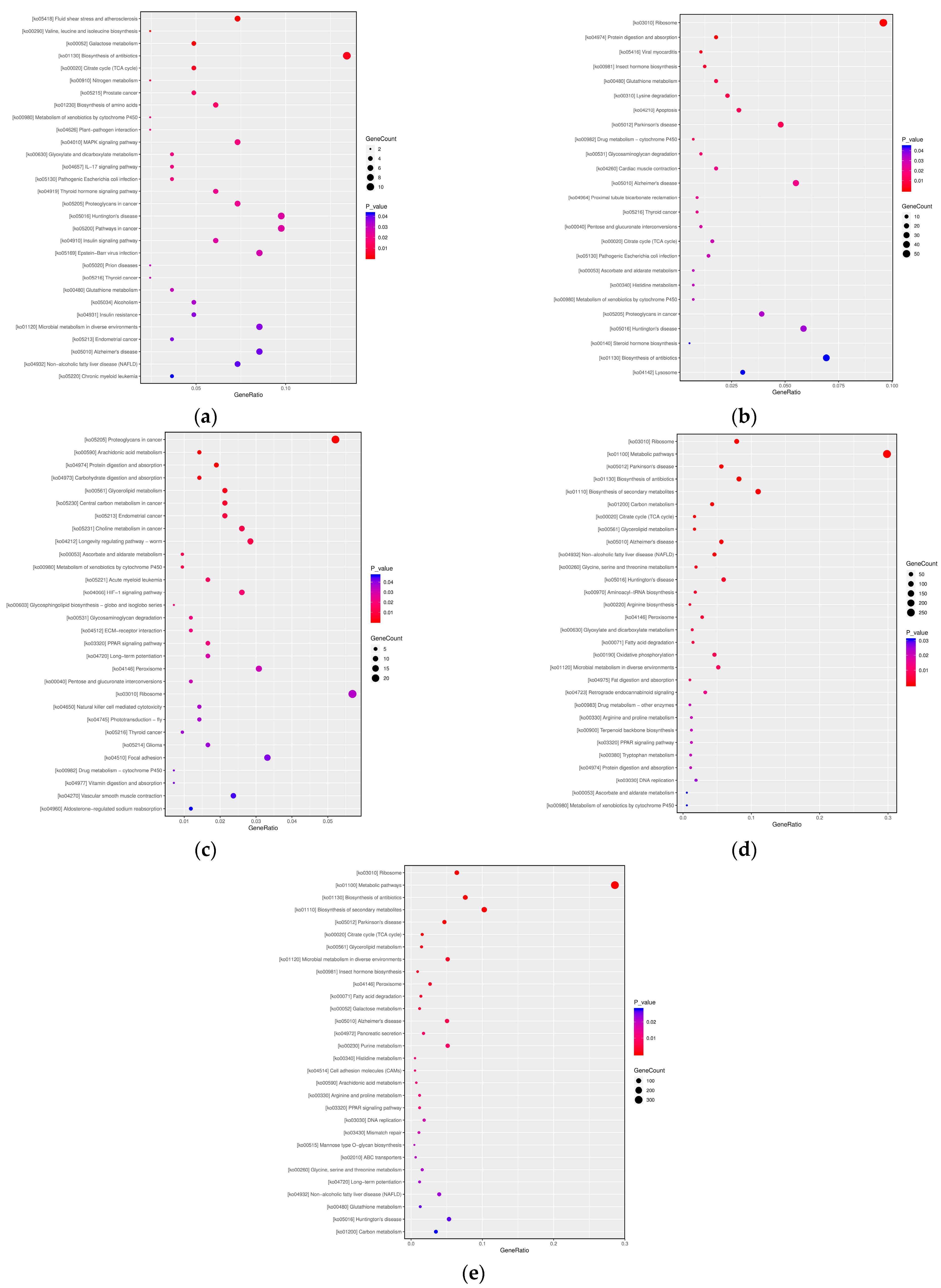
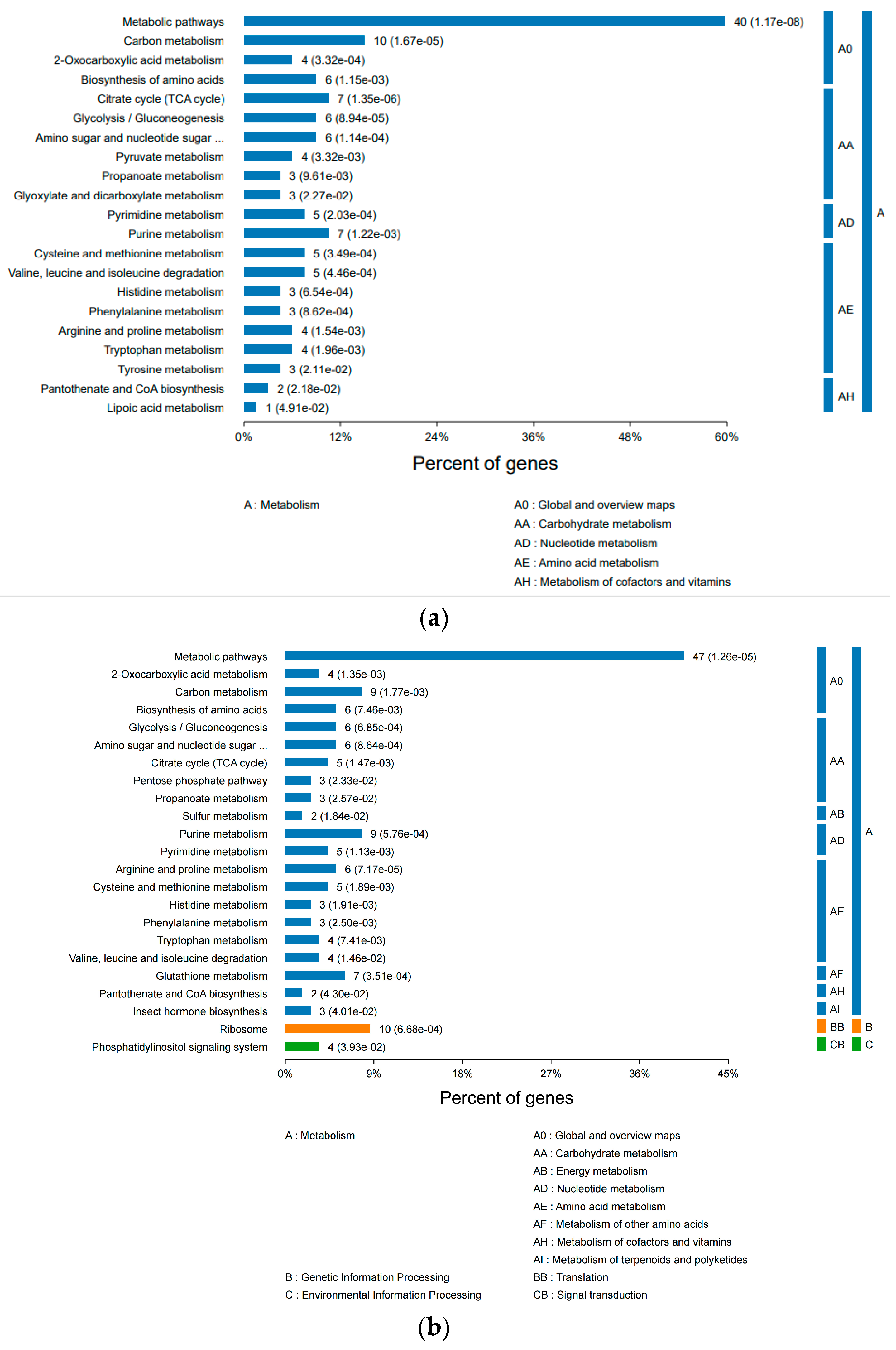
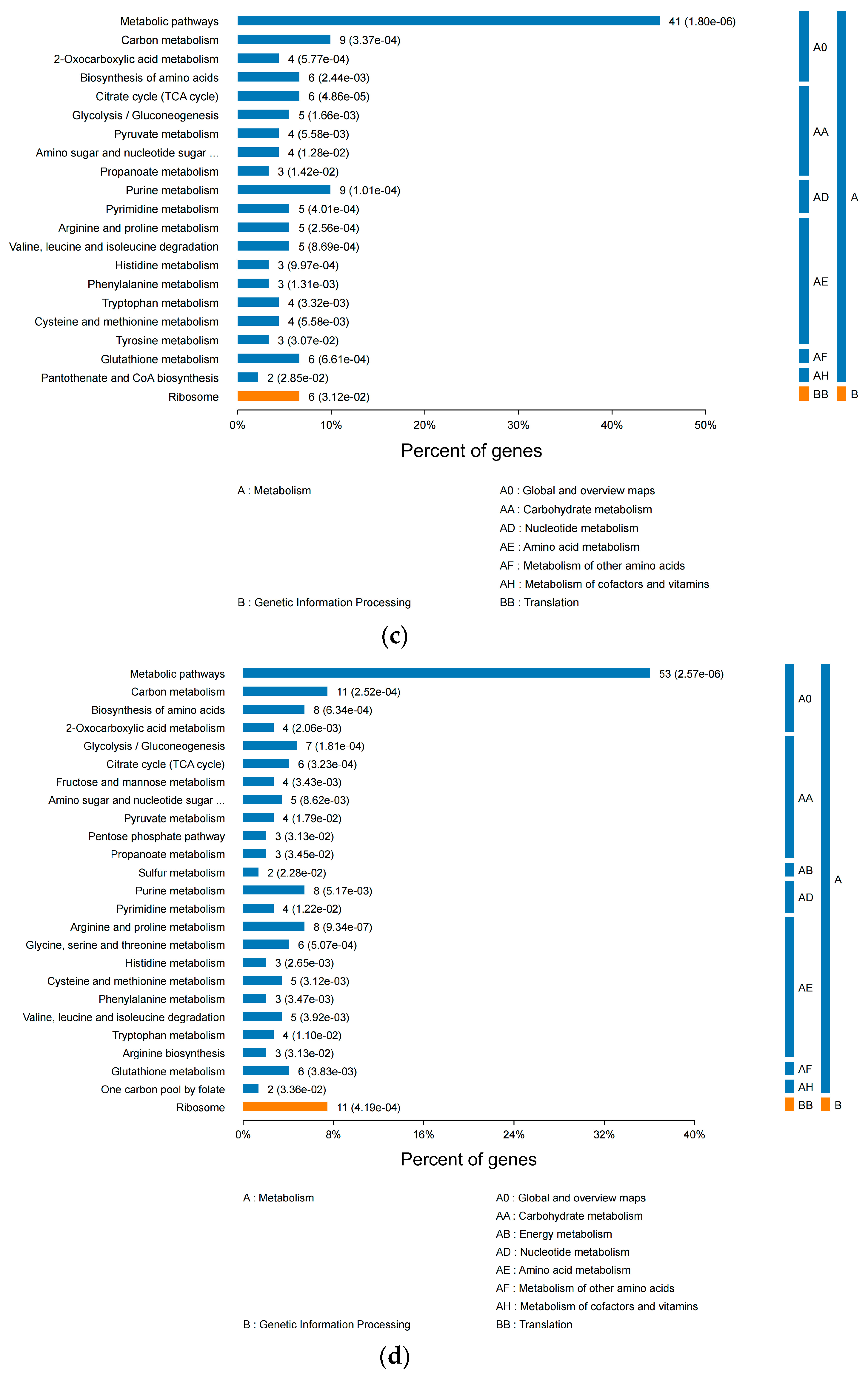
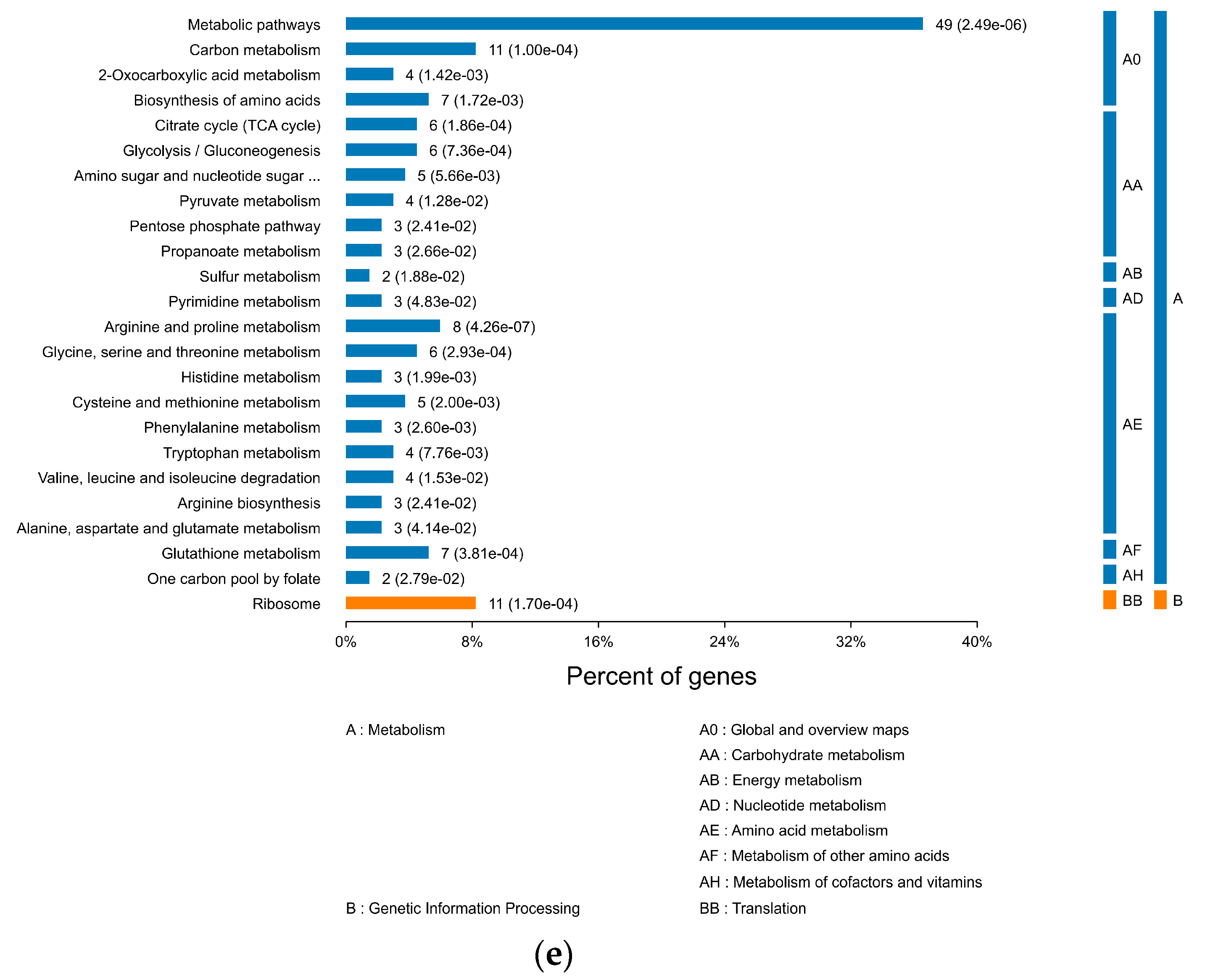
3.2.3. KEGG Pathway Analysis
3.3. Integrated Analysis of Transcriptomics and Metabolomics
4. Discussion
4.1. Metabolic Reprogramming and Nutrient Dynamics
4.2. Transcriptional Regulation of Developmental Transitions
4.3. Multi-Omics Convergence and Biological Implications
4.4. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qin, L.; Wang, X.; Li, J. Chinese Oak Silkworm Science; Chinese Science & Culture Press: Beijing, China, 2003; p. 82. [Google Scholar]
- Du, X.; Li, Y.; Wen, Z.; Li, X.; Ma, S.; Meng, N.; Mi, R.; Sun, Y.; Li, S. Research Progress on Nutritional Value and Comprehensive Utilization of Tussah Pupa. Sci. Seric. 2021, 47, 81–87. (In Chinese) [Google Scholar]
- Zhou, J.; Wink, M. Evidence for Anti-Inflammatory Activity of Isoliquiritigenin, 18β Glycyrrhetinic Acid, Ursolic Acid, and the Traditional Chinese Medicine Plants Glycyrrhiza glabra and Eriobotrya japonica, at the Molecular Level. Medicines 2019, 6, 55. [Google Scholar] [CrossRef]
- Li, Y.; Wen, Z.; Meng, N.; Li, X.; Mi, R.; Du, X. Metabolomic analyses of tussah pupa-cultivated Cordyceps militaris at different growth stages. Mycosystema 2021, 40, 1023–1038. (In Chinese) [Google Scholar]
- Meng, N.; Du, X.; Wen, Z.; Li, Y.; Ma, S.; Li, X. Study on the changes of physiological active substances and proteins during the development of tussah silkworm pupae. China Seric. 2015, 36, 24–27. (In Chinese) [Google Scholar]
- Denlinger, D.L.; Yocum, G.D.; Rinehart, J.P. Hormonal control of diapause. In Insect Endocrinology; Gilbert, L.I., Ed.; Academic Press: San Diego, CA, USA, 2012; pp. 430–463. [Google Scholar]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, F.; Nie, L. Integrating multiple ‘omics’ analysis for microbial biology: Application and methodologies. Microbiology 2010, 156, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Q.; Lin, X.; Jin, X.; Liu, L.; Wang, C.; Chen, Q.; Liu, J.; Liu, H. The use of “Omics” in lactation research in dairy cows. Int. J. Mol. Sci. 2017, 18, 983. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, H.R.; Nam, H.G. Toward systems understanding of leaf senescence: An integrated multi-omics perspective on leaf senescence research. Mol. Plant 2016, 9, 813–825. [Google Scholar] [CrossRef]
- Hettinga, K.; Zhang, L. Proteomics in Domestic Animals: From Farm to Systems Biology. In Proteomics in Domestic Animals; Springer: Berlin, Germany, 2018; pp. 463–485. [Google Scholar] [CrossRef]
- Fan, S.; Chong, J.; Ye, B.; Li, T.; Wang, T. Application of Mass Spectrometry in Silkworm Research. Biomed. Chromatogr. 2019, 33, 4476. [Google Scholar] [CrossRef]
- Chen, Q.M.; Ma, Z.G.; Wang, X.; Li, Z.Q.; Zhang, Y.; Ma, S.Y.; Zhao, P.; Xia, Q.Y. Comparative proteomic analysis of silkworm fat body after knocking out fibroin heavy chain gene: A novel insight into cross-talk between tissues. Funct. Integr. Genomics 2015, 15, 611–637. [Google Scholar] [CrossRef]
- Snyder, M.J.; Walding, J.K.; Feyereisen, R. Glutathione S-transferases from larval Manduca sexta midgut: Sequence of two cDNAs and enzyme induction. Insect Biochem. Mol. Biol. 1995, 25, 455–465. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Wang, B. A Primary Analysis on Differential Expression of Proteins in Diapause and Non-diapause Pupae of Antheraea pernyi. Sci. Seric. 2011, 37, 666–670. (In Chinese) [Google Scholar]
- Li, X.; Wang, G.; Sun, Y.; Liu, W.; He, Y.; Wang, F.; Jiang, Y.; Qin, L. Transcriptome analysis of the midgut of the Chinese oak silkworm Antheraea pernyi infected with Antheraea pernyi nucleopolyhedrovirus. PLoS ONE 2016, 11, 165959. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Chen, Z. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Arakane, Y.; Noh, M.Y.; Asano, T.; Kramer, K.J. Tyrosine metabolism for insect cuticle pigmentation and sclerotization. In Extracellular Composite Matrices in Arthropods; Cohen, E., Moussian, B., Eds.; Springer: Cham, Switzerland, 2016; pp. 165–220. [Google Scholar]
- Shao, L.; Yue, F.; Fan, J.; Su, Q.; Liu, H.; Zhang, Q.; Xu, L. Comparative proteomics and metabonomics analysis of different diapause stages revealed a new regulation mechanism of diapause in Loxostege sticticalis (Lepidoptera: Pyralidae). Molecules 2024, 29, 3472. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Mantiniotou, M.; Athanasiadis, V.; Kalompatsios, D.; Bozinou, E.; Lalas, S.I. Therapeutic capabilities of triterpenes and triterpenoids in immune and inflammatory processes: A review. Compounds 2025, 5, 2. [Google Scholar] [CrossRef]
- Li, H.; Chawla, G.; Hurlburt, A.J.; Sterrett, M.C.; Zaslaver, O.; Cox, J.; Karty, J.A.; Rosebrock, A.P.; Caudy, A.A.; Tennessen, J.M. Drosophila larvae synthesize the putative oncometabolite L-2-hydroxyglutarate during normal developmental growth. Proc. Natl. Acad. Sci. USA 2017, 114, 1353–1358. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Dermauw, W.; Van Leeuwen, T. The ABC gene family in arthropods: Comparative genomics and role in insecticide transport and resistance. Insect Biochem. Mol. Biol. 2014, 45, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, U.; Yukari, N.; Akihiro, N.; Hidetsugu, T.; Anirban, C.; Kunio, I.; Naoya, K. Ribosomal protein gene knockdown causes developmental defects in zebrafish. PLoS ONE 2006, 1, 37. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species? Biochem. J. 2009, 417, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Li, X.; Tian, Y. Mitochondrial-to-nuclear communication in aging: An epigenetic perspective. Trends Biochem. Sci. 2022, 47, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef]
- Ramnarine, T.J.S.; Grath, S.; Parsch, J. Natural variation in the transcriptional response of Drosophila melanogaster to oxidative stress. Genes Genomes Genet. 2021, 12, jkab366. [Google Scholar] [CrossRef]
- Gegner, J.; Vogel, H.; Billion, A.; Förster, F.; Vilcinskas, A. Complete metamorphosis in Manduca sexta involves specific changes in DNA methylation patterns. Front. Ecol. Evol. 2021, 9, 646281. [Google Scholar] [CrossRef]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef]
- Yamamoto, K.; Zhang, P.; Miake, F.; Kashige, N.; Aso, Y.; Banno, Y.; Fujii, H. Cloning, expression and characterization of theta-class glutathione S-transferase from the silkworm, Bombyx mori. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2005, 141, 340–346. [Google Scholar] [CrossRef]
- Riddiford, L.M. How does juvenile hormone control insect metamorphosis and reproduction? Gen. Comp. Endocrinol. 2012, 179, 477–484. [Google Scholar] [CrossRef]
- Teleman, A.A.; Chen, Y.W.; Cohen, S.M. Drosophila melted modulates FoxO and TOR activity. Dev. Cell 2005, 9, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Teixeira, F.K.; Czech, B.; Preall, J.; Lehmann, R. Regulation of ribosome biogenesis and protein synthesis controls germline stem cell differentiation. Cell Stem Cell 2016, 18, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Rösner, J.; Merzendorfer, H. Transcriptional plasticity of different ABC transporter genes from Tribolium castaneum contributes to diflubenzuron resistance. Insect Biochem. Mol. Biol. 2020, 116, 103282. [Google Scholar] [CrossRef]
- Villani, S.; Calcagnile, M.; Demitri, C.; Alifano, P. Galleria mellonella (Greater Wax Moth) as a reliable animal model to study the efficacy of nanomaterials in fighting pathogens. Nanomaterials 2025, 15, 67. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Number | Proportion |
|---|---|---|
| Organic acids and derivatives | 135 | 24.46% |
| Organic heterocyclic compounds | 102 | 18.48% |
| Lipids and lipid-like molecules | 73 | 13.22% |
| Organic oxygen-containing compounds | 64 | 11.59% |
| Phenylpropanoids and polyketides | 63 | 11.41% |
| Benzene-type compounds | 60 | 10.87% |
| Nucleosides, nucleotides, and derivatives | 32 | 5.80% |
| Alkaloids and derivatives | 7 | 1.27% |
| Organic nitrogen compounds | 7 | 1.27% |
| Lignans, neolignans, and related compounds | 5 | 0.91% |
| Homogeneous non-metal compounds | 2 | 0.36% |
| Derivatives of hydrocarbons | 1 | 0.18% |
| Organic sulfur compounds | 1 | 0.18% |
| Group | Total | Upregulated | Downregulated |
|---|---|---|---|
| T1 vs. CK | 424 | 146 | 278 |
| T2 vs. CK | 509 | 243 | 266 |
| T3 vs. CK | 564 | 276 | 288 |
| T4 vs. CK | 647 | 356 | 291 |
| T5 vs. CK | 710 | 411 | 299 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Sun, Y.; Li, Y.; Li, X.; Wen, Z.; Mi, R.; Meng, N.; Du, X. Integrated Multi-Omics Analysis of the Developmental Stages of Antheraea pernyi Pupae: Dynamic Changes in Metabolite Profiles and Gene Expression. Insects 2025, 16, 745. https://doi.org/10.3390/insects16070745
Ma S, Sun Y, Li Y, Li X, Wen Z, Mi R, Meng N, Du X. Integrated Multi-Omics Analysis of the Developmental Stages of Antheraea pernyi Pupae: Dynamic Changes in Metabolite Profiles and Gene Expression. Insects. 2025; 16(7):745. https://doi.org/10.3390/insects16070745
Chicago/Turabian StyleMa, Shuhui, Yongxin Sun, Yajie Li, Xuejun Li, Zhixin Wen, Rui Mi, Nan Meng, and Xingfan Du. 2025. "Integrated Multi-Omics Analysis of the Developmental Stages of Antheraea pernyi Pupae: Dynamic Changes in Metabolite Profiles and Gene Expression" Insects 16, no. 7: 745. https://doi.org/10.3390/insects16070745
APA StyleMa, S., Sun, Y., Li, Y., Li, X., Wen, Z., Mi, R., Meng, N., & Du, X. (2025). Integrated Multi-Omics Analysis of the Developmental Stages of Antheraea pernyi Pupae: Dynamic Changes in Metabolite Profiles and Gene Expression. Insects, 16(7), 745. https://doi.org/10.3390/insects16070745