
Functional Differentiation Reconfiguration in the Midgut of Nezara viridula (Hemiptera: Pentatomidae) Based on Transcriptomics: Multilayer Enrichment Analysis and Topological Network Interpretation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Transcriptome Data Preprocessing and Functional Annotation Pipeline
2.2. Pathway Activity and Segmental Transcriptomic Analysis
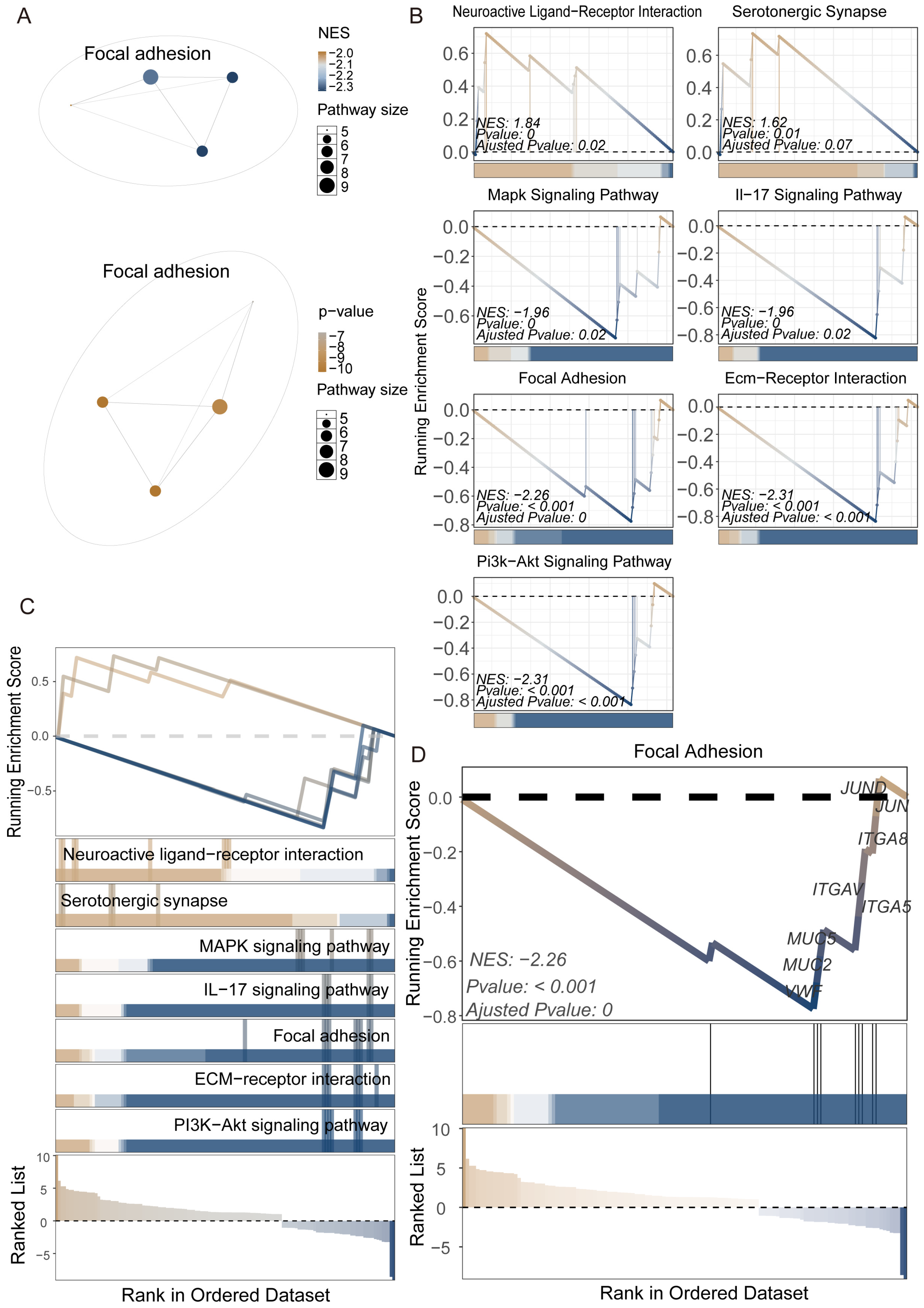
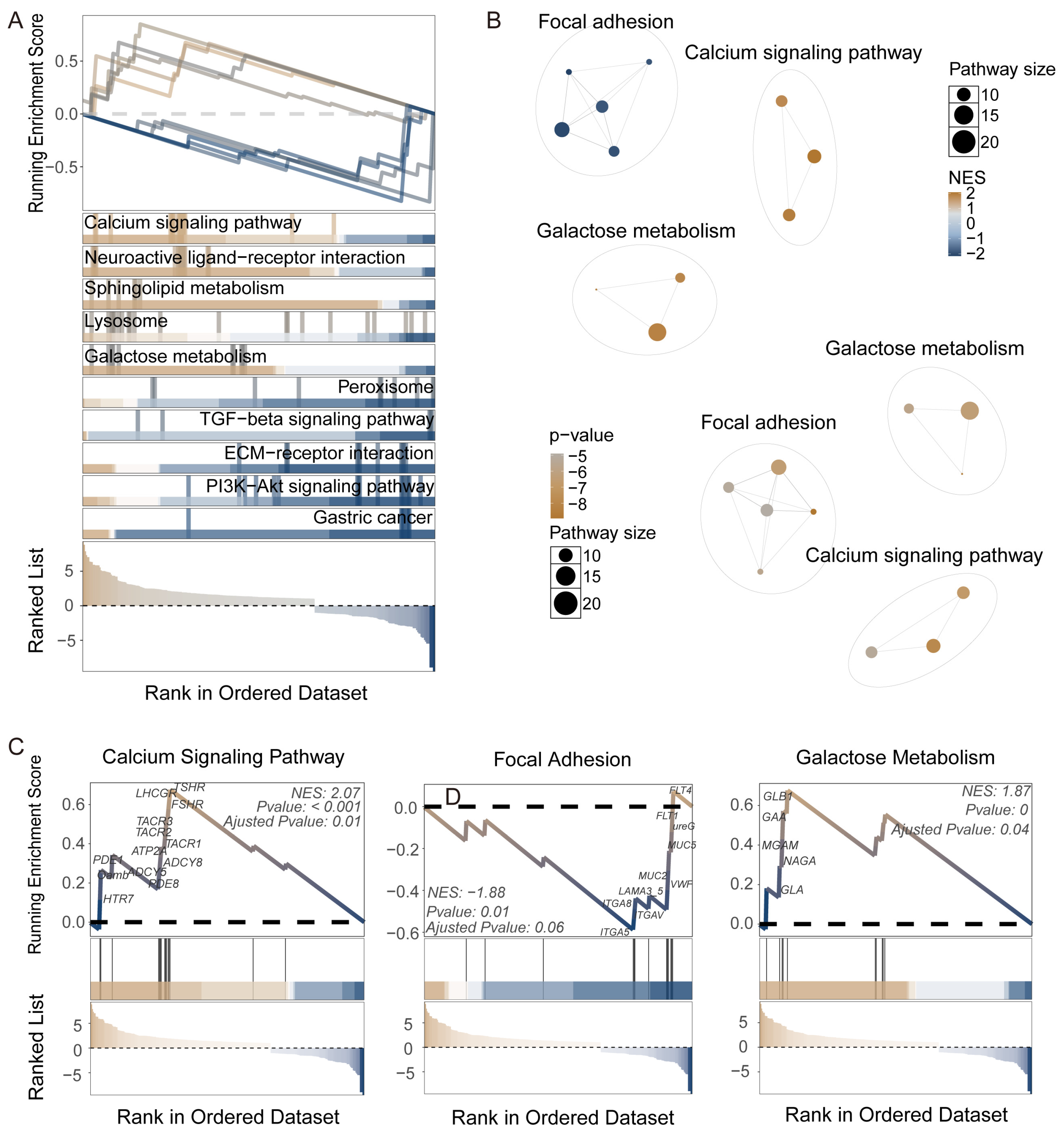
2.3. Advanced Visualization of Gene Set Enrichment Analysis
2.4. Topological Mapping of Gene Interaction Networks
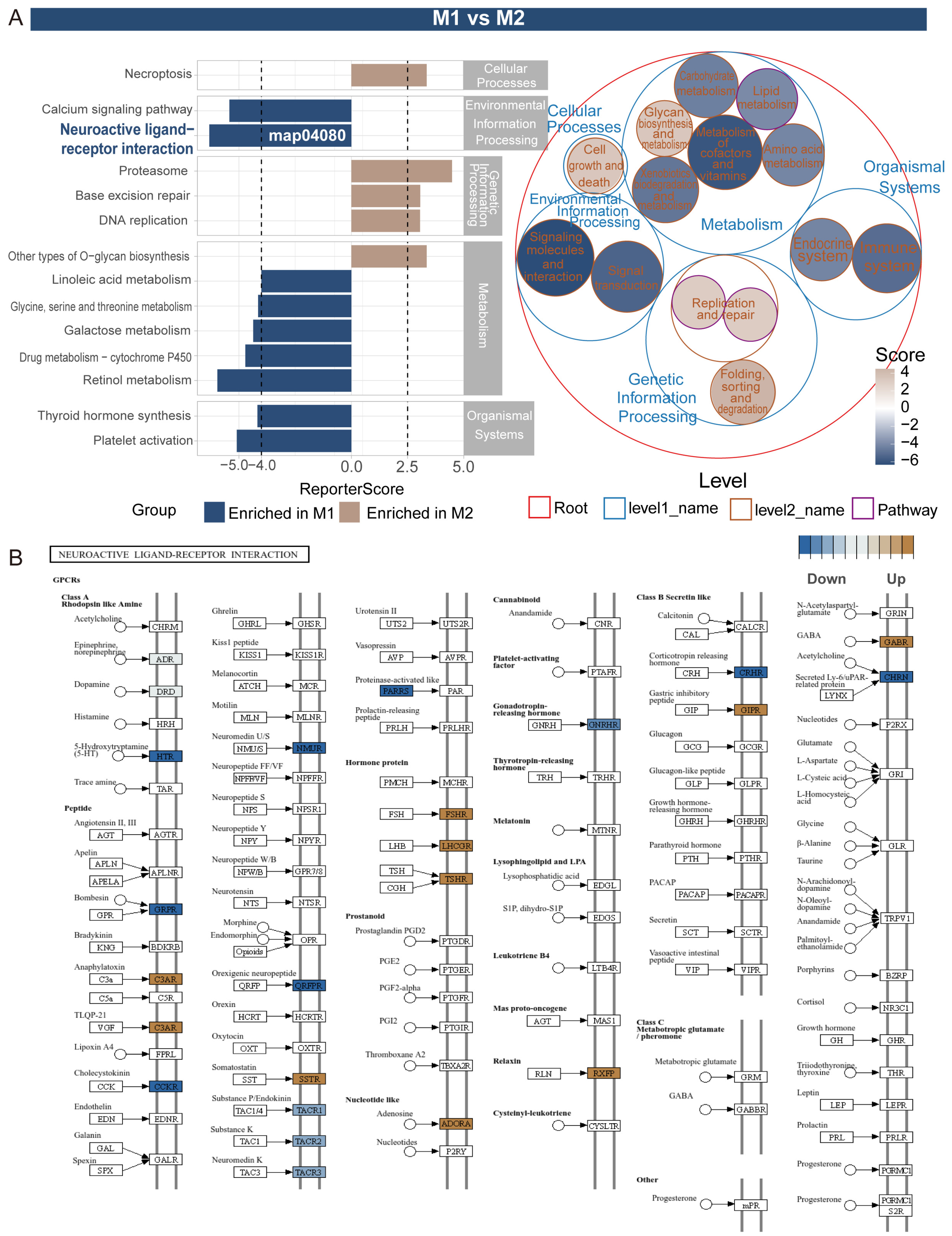
2.5. Pathway Activity Differentiation Analysis Across Tissues Using ReporterScore
3. Results
3.1. PCA of Different Midgut Regions in Nezara viridula
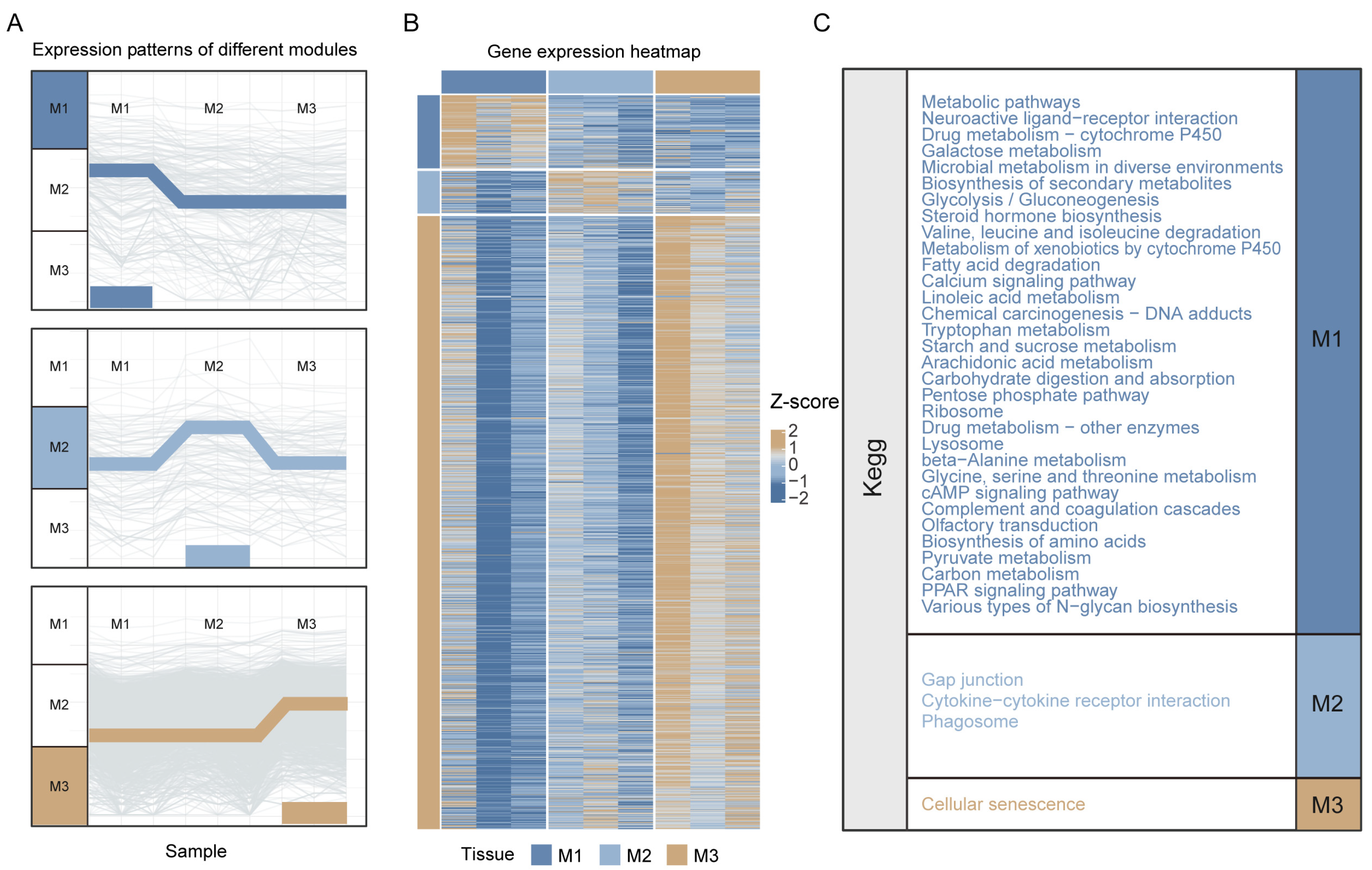
3.2. Enrichment Analysis of Highly Expressed Genes Specific to Different Midgut Regions
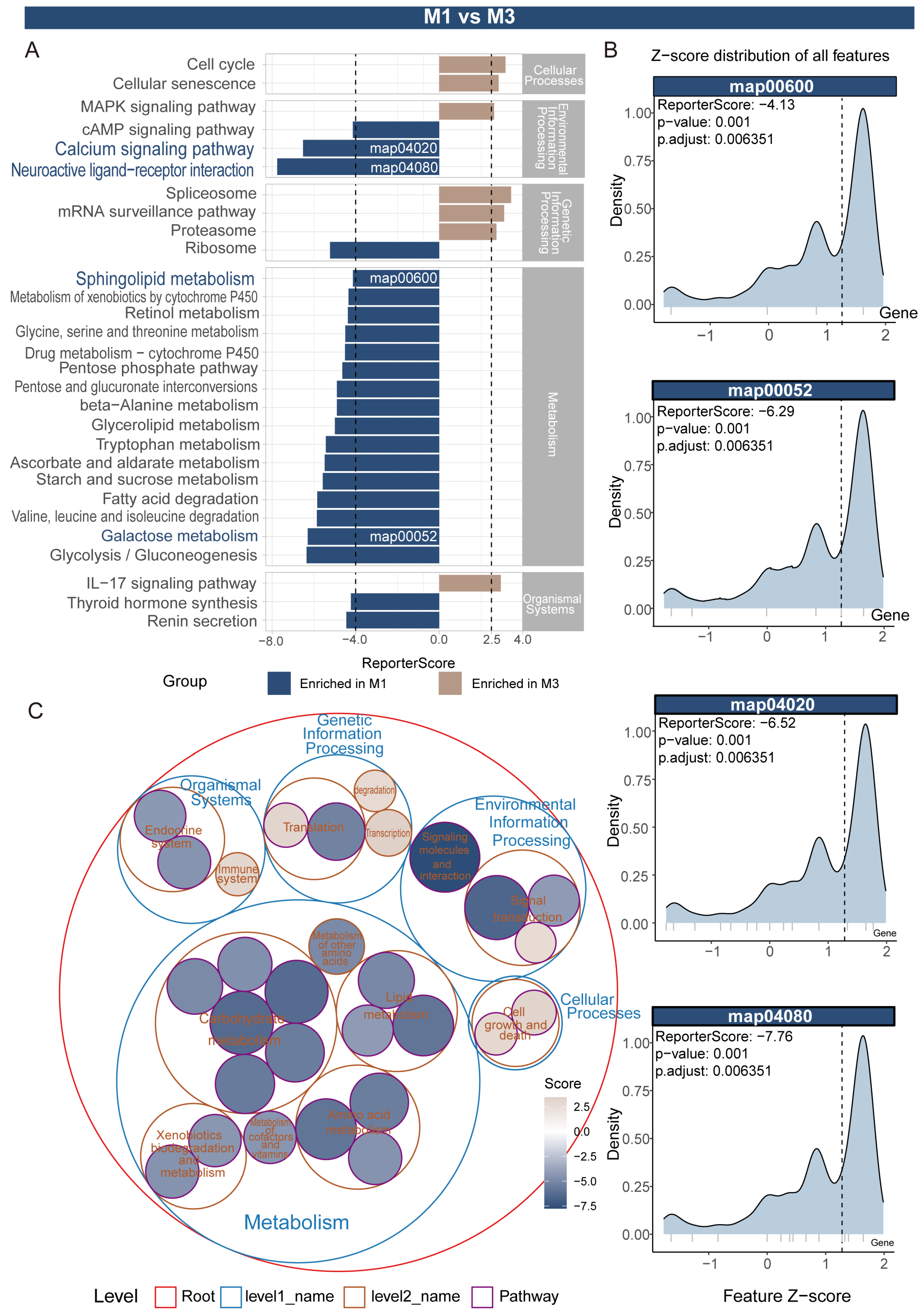
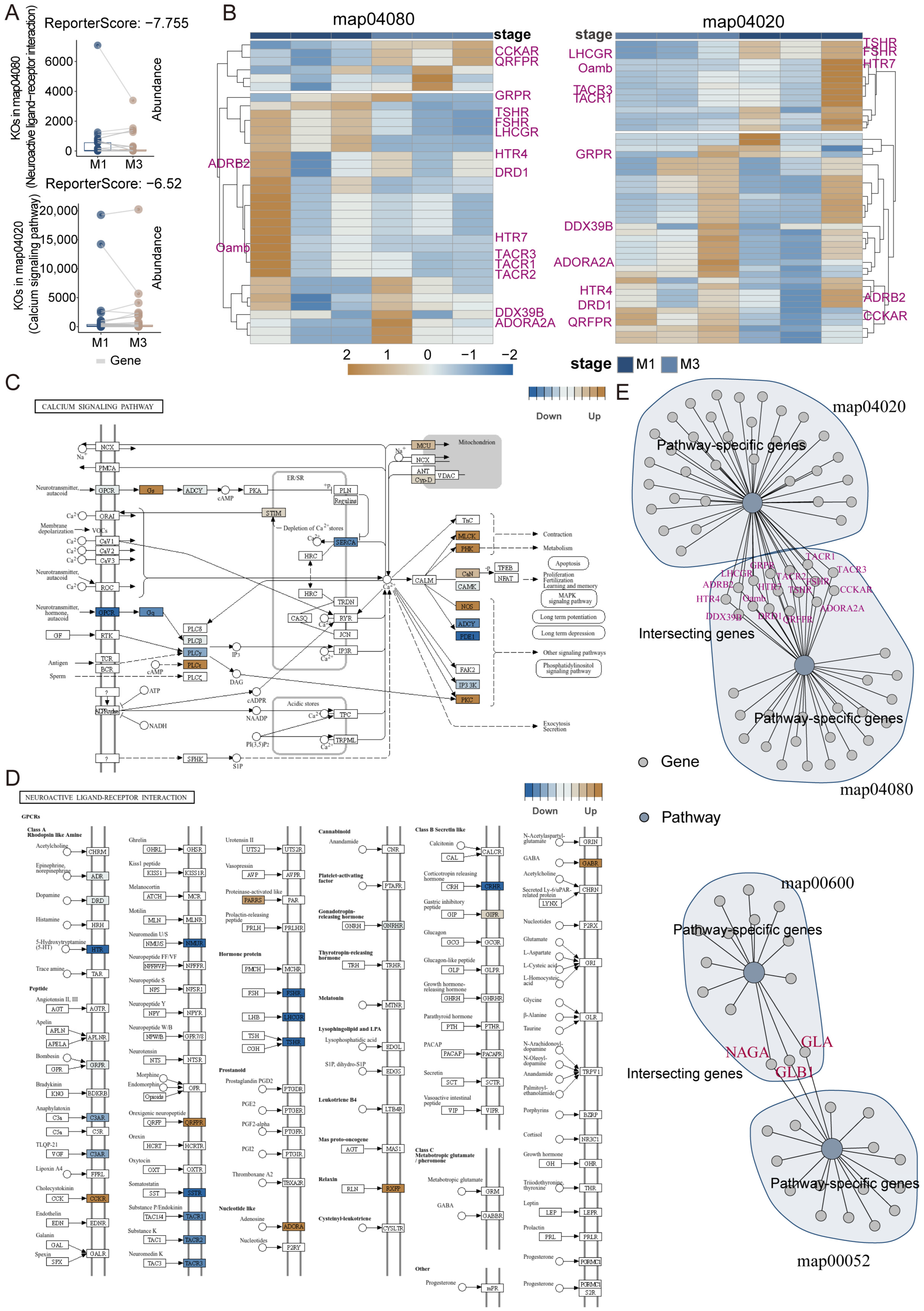
3.3. Construction of KEGG Pathway Network and Systematic Identification of Functional Modules in Nezara viridula
3.4. Gene Regulatory System Analysis and GRSA Visualization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Holtof, M.; Lenaerts, C.; Cullen, D.; Vanden Broeck, J. Extracellular nutrient digestion and absorption in the insect gut. Cell Tissue Res. 2019, 377, 397–414. [Google Scholar] [CrossRef]
- Caccia, S.; Casartelli, M.; Tettamanti, G. The amazing complexity of insect midgut cells: Types, peculiarities, and functions. Cell Tissue Res. 2019, 377, 505–525. [Google Scholar] [CrossRef]
- Huang, J.-H.; Jing, X.; Douglas, A.E. The multi-tasking gut epithelium of insects. Insect Biochem. Mol. Biol. 2015, 67, 15–20. [Google Scholar] [CrossRef]
- Lemaitre, B.; Miguel-Aliaga, I. The Digestive Tract of Drosophila melanogaster. Annu. Rev. Genet. 2013, 47, 377–404. [Google Scholar] [CrossRef]
- Franzetti, E.; Casartelli, M.; D’Antona, P.; Montali, A.; Romanelli, D.; Cappellozza, S.; Caccia, S.; Grimaldi, A.; De Eguileor, M.; Tettamanti, G. Midgut epithelium in molting silkworm: A fine balance among cell growth, differentiation, and survival. Arthropod Struct. Dev. 2016, 45, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Tillman, P.G. Composition and Abundance of Stink Bugs (Heteroptera: Pentatomidae) in Corn. Environ. Entomol. 2010, 39, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Hirose, E.; Panizzi, A.R.; De Souza, J.T.; Cattelan, A.J.; Aldrich, J.R. Bacteria in the Gut of Southern Green Stink Bug (Heteroptera: Pentatomidae). Ann. Entomol. Soc. Am. 2006, 99, 91–95. [Google Scholar] [CrossRef]
- Denecke, S.; Ioannidis, P.; Buer, B.; Ilias, A.; Douris, V.; Topalis, P.; Nauen, R.; Geibel, S.; Vontas, J. A transcriptomic and proteomic atlas of expression in the Nezara viridula (Heteroptera:Pentatomidae) midgut suggests the compartmentalization of xenobiotic metabolism and nutrient digestion. BMC Genom. 2020, 21, 129. [Google Scholar] [CrossRef]
- Cantón, P.E.; Bonning, B.C. Transcription and Activity of Digestive Enzymes of Nezara viridula Maintained on Different Plant Diets. Front. Physiol. 2020, 10, 1553. [Google Scholar] [CrossRef]
- Cantón, P.E.; Bonning, B.C. Proteases and nucleases across midgut tissues of Nezara viridula (Hemiptera:Pentatomidae) display distinct activity profiles that are conserved through life stages. J. Insect Physiol. 2019, 119, 103965. [Google Scholar] [CrossRef]
- Dias, R.O.; Cardoso, C.; Pimentel, A.C.; Damasceno, T.F.; Ferreira, C.; Terra, W.R. The roles of mucus-forming mucins, peritrophins and peritrophins with mucin domains in the insect midgut. Insect Mol. Biol. 2018, 27, 46–60. [Google Scholar] [CrossRef]
- Terra, W.R.; Ferreira, C. Biochemistry and Molecular Biology of Digestion; Elsevier: Amsterdam, The Netherlands, 2012; pp. 365–418. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Patil, K.R.; Nielsen, J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. USA 2005, 102, 2685–2689. [Google Scholar] [CrossRef]
- Tjeldnes, H.; Labun, K.; Torres Cleuren, Y.; Chyżyńska, K.; Świrski, M.; Valen, E. ORFik: A comprehensive R toolkit for the analysis of translation. BMC Bioinform. 2021, 22, 336. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novoassembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Thirteen years of clusterProfiler. Innovation 2024, 5, 100722. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Tao, W.; Zhou, J. GseaVis: An R Package for Enhanced Visualization of Gene Set Enrichment Analysis in Biomedicine. Med. Res. 2025. [Google Scholar] [CrossRef]
- Kerseviciute, I.; Gordevicius, J. aPEAR: An R package for autonomous visualization of pathway enrichment networks. Bioinformatics 2023, 39, btad672. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Peng, C.; Chen, Q.; Tan, S.; Shen, X.; Jiang, C. Generalized reporter score-based enrichment analysis for omics data. Brief. Bioinform. 2024, 25, bbae116. [Google Scholar] [CrossRef]
- Veenstra, J.A.; Agricola, H.-J.; Sellami, A. Regulatory peptides in fruit fly midgut. Cell Tissue Res. 2008, 334, 499–516. [Google Scholar] [CrossRef]
- Audsley, N.; Weaver, R.J. Neuropeptides associated with the regulation of feeding in insects. Gen. Comp. Endocrinol. 2009, 162, 93–104. [Google Scholar] [CrossRef]
- Nässel, D.R.; Zandawala, M. Recent advances in neuropeptide signaling in Drosophila, from genes to physiology and behavior. Prog. Neurobiol. 2019, 179, 101607. [Google Scholar] [CrossRef]
- Dolezelova, E.; Nothacker, H.-P.; Civelli, O.; Bryant, P.J.; Zurovec, M. A Drosophila adenosine receptor activates cAMP and calcium signaling. Insect Biochem. Mol. Biol. 2007, 37, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Hakim, R.S.; Baldwin, K.; Smagghe, G. Regulation of Midgut Growth, Development, and Metamorphosis. Annu. Rev. Entomol. 2010, 55, 593–608. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, D.; Liang, J.; Bu, W. Functional Differentiation Reconfiguration in the Midgut of Nezara viridula (Hemiptera: Pentatomidae) Based on Transcriptomics: Multilayer Enrichment Analysis and Topological Network Interpretation. Insects 2025, 16, 634. https://doi.org/10.3390/insects16060634
Yu D, Liang J, Bu W. Functional Differentiation Reconfiguration in the Midgut of Nezara viridula (Hemiptera: Pentatomidae) Based on Transcriptomics: Multilayer Enrichment Analysis and Topological Network Interpretation. Insects. 2025; 16(6):634. https://doi.org/10.3390/insects16060634
Chicago/Turabian StyleYu, Dongyue, Jingyu Liang, and Wenjun Bu. 2025. "Functional Differentiation Reconfiguration in the Midgut of Nezara viridula (Hemiptera: Pentatomidae) Based on Transcriptomics: Multilayer Enrichment Analysis and Topological Network Interpretation" Insects 16, no. 6: 634. https://doi.org/10.3390/insects16060634
APA StyleYu, D., Liang, J., & Bu, W. (2025). Functional Differentiation Reconfiguration in the Midgut of Nezara viridula (Hemiptera: Pentatomidae) Based on Transcriptomics: Multilayer Enrichment Analysis and Topological Network Interpretation. Insects, 16(6), 634. https://doi.org/10.3390/insects16060634