2. Materials and Methods
2.1. Video Assays
For video assay, flies were gently knocked into 4 dram (~15 mL) glass vials, using 6 flies per vial and 4 vials per group, and each vial was covered with a glass coverslip. Each vial was individually placed in a dark box illuminated using two LED lights, and two synchronized video cameras were used to capture fluorescence in 4-min videos (see
Supplementary Figure S1). Prior to recording, flies were transferred to media supplemented with 200 µg/mL mifepristone for the Gene-Switch system or 640 µg/mL doxycycline for the Tet-ON system to initiate fluorescent protein expression and then removed from the drug media after 24 or 48 h, as indicated. Recordings were taken immediately after removal from the drug (day 1) and every day over a period of 5–6 days. Videos were analyzed using FluoreScore version 1 software, which identifies each fly and quantifies pixel intensity in the area of the flies in each frame [
9]. The data from the cameras was combined to yield a fluorescence intensity value for each vial at each time point, and average fluorescence intensity values for the 4 vials in each group were averaged. Fluorescence typically peaked at 24 h after cessation of drug treatment for the Gene-Switch system and at 4–5 days after cessation of drug treatment for the Tet-ON system, after which it decreased exponentially. The natural logarithm of each average fluorescence value was taken, the values for each group were plotted as ln(fluorescence) versus time, and the slope of the resulting linear regression from peak to minimum was used to calculate half-life.
2.2. Fly Culture and Strains
Drosophila melanogaster were cultured at 25 °C using a standard agar/dextrose/cornmeal/yeast media [
10], and adult flies were passaged to fresh media every other day.
Actin-GS-255B is previously described and contains the
Actin5C gene promoter driving tissue-general expression of Gene-Switch [
1]. The
repo-GS strain drives Gene-Switch expression in glial cells and was generously provided by Amita Sehgal [
11]. The
5966-GS strain expresses Gene-Switch in midgut enterocytes and was generously provided by Pankaj Kapahi [
12].
Actin88F-GS drives expression of Gene-Switch in flight muscle and was generously provided by Jason Karpac [
13]. The
w[1118];
3xP3-GFP[M1] strain transgenic construct includes a synthetic promoter containing three binding sites for the Eyeless/PAX6 transcription factor and was generously provided by Ernst Wimmer [
14]; the construct drives high-level expression of eGFP specifically in retinal tissue and is abbreviated here as
eyeless-GFP. Certain strains were obtained from the Bloomington Drosophila Stock Center (BDSC), including strain
y[1] w[*]; P{w[+mC] = elav-Switch.O}GSG301 (BDSC#43642), which is abbreviated here as
y; Elav-GS, strain
y[1] w[*]; P{tubP-GAL4}LL7/TM3 (BDSC#5138), abbreviated here as
tub-GAL4, strain
y[1] w[*]; wg[Sp-1]/CyO;
P{Mhc-Switch.O}GSG314-2 (BDSC#43641), abbreviated here as
Mhc-GS, strain
w[1118]; P{UAS-MitoTimer}3 (BDSC#57323), abbreviated here as
UAS-MT [
15,
16], and strain
y[1] w[*];
wg[Sp-1]/CyO,
P{Wee-P.ph0}Bacc[Wee-P20];
P{y[+t7.7] w[+mC] = 20XUAS-6XmCherry-HA}attP2 (BDSC#52268), abbreviated here as
UAS-mCherry. The
w[1118] strain (BDSC#5905) is the isogenized version (
w[1118]-iso;
2-iso;
3-iso) that was previously cured of Wolbachia [
10]. The
UAS-eGFP strain employed is abbreviated as
Ultra-GFP and contains multiple copies of a
UAS-2xeGFP construct on both the second and third chromosomes and has been previously described [
1]. To generate mated females, age-synchronized virgin females at 1–2 days of age were combined with young (3–14 day old) male
w[1118] flies in regular food vials, with 20 females and 20 males per vial, for 48 h, after which the males were removed.
2.3. Generation of Multi-Copy Strains
Generation of Tet-ON strain REDA: The Tet-ON system driver construct rtTA consists of the
Actin5C promoter driving expression of the reverse tetracycline trans-activator protein called rtTA [
1]. The construct is inserted on the third chromosome, and the insertion strain is named
rtTA(3)E2. The target construct contains a synthetic promoter consisting of seven Tet-operator (TetO) sequences fused to the core promoter sequences of the
hsp70 gene, driving expression of DsRED [
17]. Two independent strains bearing insertions of this
TetO-DsRED construct on the third chromosome were used,
TetO-DsRED[6] and
TetO-DsRED[26B] [
17], each in the
w[1118] genetic background. The target transgenic constructs bear the
mini-white+ marker gene and have light orange-colored eyes resulting from partial rescue of the
w[1118] mutant phenotype. To increase the copy number of the target constructs, each strain was crossed to
P(ry+,Δ2-3)99B transposase strain [
18], and third chromosomes were isolated that had increased dosage of the
mini-white+ marker, as indicated by a more red eye color, and named
TetO-DsRED[6-7] and
TetO-DsRED[26B-6], respectively. The increased copy number of the constructs was confirmed by crossing to the
rtTA(3)E2 strain, assaying progeny ± DOX treatment, and confirming a total two-fold increase in DsRED signal using fluorescence microscopy. The
TetO-DsRED[6-7] and
TetO-DsRED[26B-6] insertions were then recombined onto the same third chromosome, and the increased copy number was confirmed by again crossing to the
rtTA(3)E2 strain, assaying progeny ± DOX treatment, and confirming a total four-fold increase in DsRED signal using fluorescence microscopy. Finally, the chromosome bearing the
TetO-DsRED[6-7] and
TetO-DsRED[26B-6] insertions was recombined with
rtTA(3)E2 to generate a chromosome bearing all three insertions on the same third chromosome, balanced over
TM3 Sb balancer, and in a
y[*] w[*] genetic background; the resulting strain
y[*] w[*];
TetO-DsRED[6-7],
TetO-DsRED[26B-6], rtTA(3)E2/TM3 Sb, which is abbreviated as “REDA”. The yellow mutant body color allows for increased detection of fluorescence from internal tissues relative to a wild-type body color.
Multi-copy mitoGFP strain: The strains
P{UAS-mito-HA-GFP.AP}2 and
P{UAS-mito-HA-GFP.AP}3 [
19] were obtained from BDSC (BDSC#8442; BDSC#8443). Each strain was crossed to
P(ry+,Δ2-3)99B transposase strain, and chromosomes were isolated that had increased dosage of the
mini-white+ marker strain, as indicated by a more red eye color, and named
UAS-mitoGFP[AP2-4] and
UAS-mitoGFP[AP3-7], respectively. The increased copy number of the constructs was confirmed by crossing to the
tub-GAL4 strain and assaying progeny to confirm a two-fold increase in GFP signal using fluorescence microscopy.
UAS-mitoGFP[AP2-4] and
UAS-mitoGFP[AP3-7] were then combined into the same strain using double-balancer crosses to yield strain
y[*] w[*]; UAS-mitoGFP[AP2-4]; UAS-mitoGFP[AP3-7]/TM3 Ser.
Multi-copy MitoTimer strain: The
UAS-MT strain was crossed to the
P(ry+,Δ2-3)99B transposase strain, and a third chromosome was isolated that had an increased dosage of the
mini-white+ marker, as indicated by a more red eye color, and named
UAS-MT[1]. The increased copy number of the construct was confirmed by crossing to the
tub-GAL4 strain and assaying progeny to confirm a two-fold increase in green and red fluorescence signals using fluorescence microscopy. The
UAS-MT[1] was crossed to the
P(ry+,Δ2-3)99B transposase strain, and the third chromosome was isolated that had an increased dosage of the
mini-white+ marker strain, as indicated by a more red eye color, and named
UAS-MT[1-1]. The increased copy number of the construct was confirmed by crossing to the
tub-GAL4 strain and assaying progeny to confirm a three-fold increase in green and red fluorescence signals using fluorescence microscopy (
Figure S5).
2.4. Cameras and Filters
The 3D tracking and quantification of GFP fluorescence were carried out essentially as previously described [
9]. Briefly, two video cameras (Grasshopper type GRAS-20S4C, Point Grey Research, Scottsdale, AZ, USA) were directed at the glass vial containing the fly. The vial was illuminated by two LED lights, and video was recorded at 30 fps. For GFP fluorescence tracking, blue LEDs were used as follows: The MF469-35 filters (Thorlabs, Inc., Newton, NJ, USA) were fixed in front of the blue LEDs to limit the light to a range of approximately 452 nm to 486 nm, which overlaps the eGFP absorption peak at approximately 488 nm. The MF525-39 filters were placed in front of the camera lenses to limit the light to the range of approximately 510 nm to 548 nm, which overlaps the emission peak for eGFP at approximately 509 nm. For DsRED tracking, green LEDs were used as follows: The TRITC excitation filters (MF542-20; Thorlabs, Inc.) were fixed in front of the green LEDs to limit the light to a range of approximately 532 nm to 552 nm, which overlaps the DsRED absorption peak at approximately 555 nm. The TRITC/CY3.5 emission filters (Thorlabs MF620-52) were placed in front of the camera lenses to limit the light to the range of approximately 595 nm to 645 nm, which overlaps the emission peak for DsRED at approximately 590 nm.
2.5. Vials, Media and Fly Handling for Video Assays
Assay of tissue-general eGFP in parallel in young and old, male and female flies. Flies were maintained as adults on NutriFly Grape Agar Premix media (Genesee Scientific 47-102), adjusted to 0.5% Casein protein (Sigma/Aldrich, St. Louis, MO, USA); this is to reduce background green fluorescence from the gut that can result from the standard Drosophila media. For assay, 6 flies were knocked into an empty 4 dram glass shell vial (Kimble 60965-4, Wayne, PA, USA), without anesthetization, on a foam mouse pad to avoid damage to vials. The bottom of the vial was covered with a round piece of black filter paper to reduce glare, and the top of the vial was covered with a glass coverslip.
Assay of eGFP, mitoGFP and DsRED in various tissue patterns. Clear glass shell vials (4 mL; ThermoFisher, Waltham, MA, USA) were filled with 500 µL media. The media was NutriFly Grape Agar Premix (Genesee Scientific 47-102, San Diego, CA, USA), adjusted to 0.5% Casein protein (Sigma/Aldrich) and adjusted to 0.25% wgt/vol blue dye #1. This media was used to reduce background fluorescence. The media was adjusted to a final concentration of 200 µg/mL mifepristone for the Gene-Switch system or to 640 µg/mL doxycycline for the Tet-ON system. To introduce flies, the flies were lightly anesthetized with CO2 gas, knocked into the vial, and the vial covered at the top with a small piece of clear tape (Grey Parrot brand crystal clear office tape) to prevent flies from escaping without compromising visibility. Flies were passaged to fresh vials every other day (using light CO2 anesthesia). When not being assayed, all flies were kept in an incubator at 25 °C, with the exception of mitoGFP flies, which were maintained at 22 (±0.5) °C.
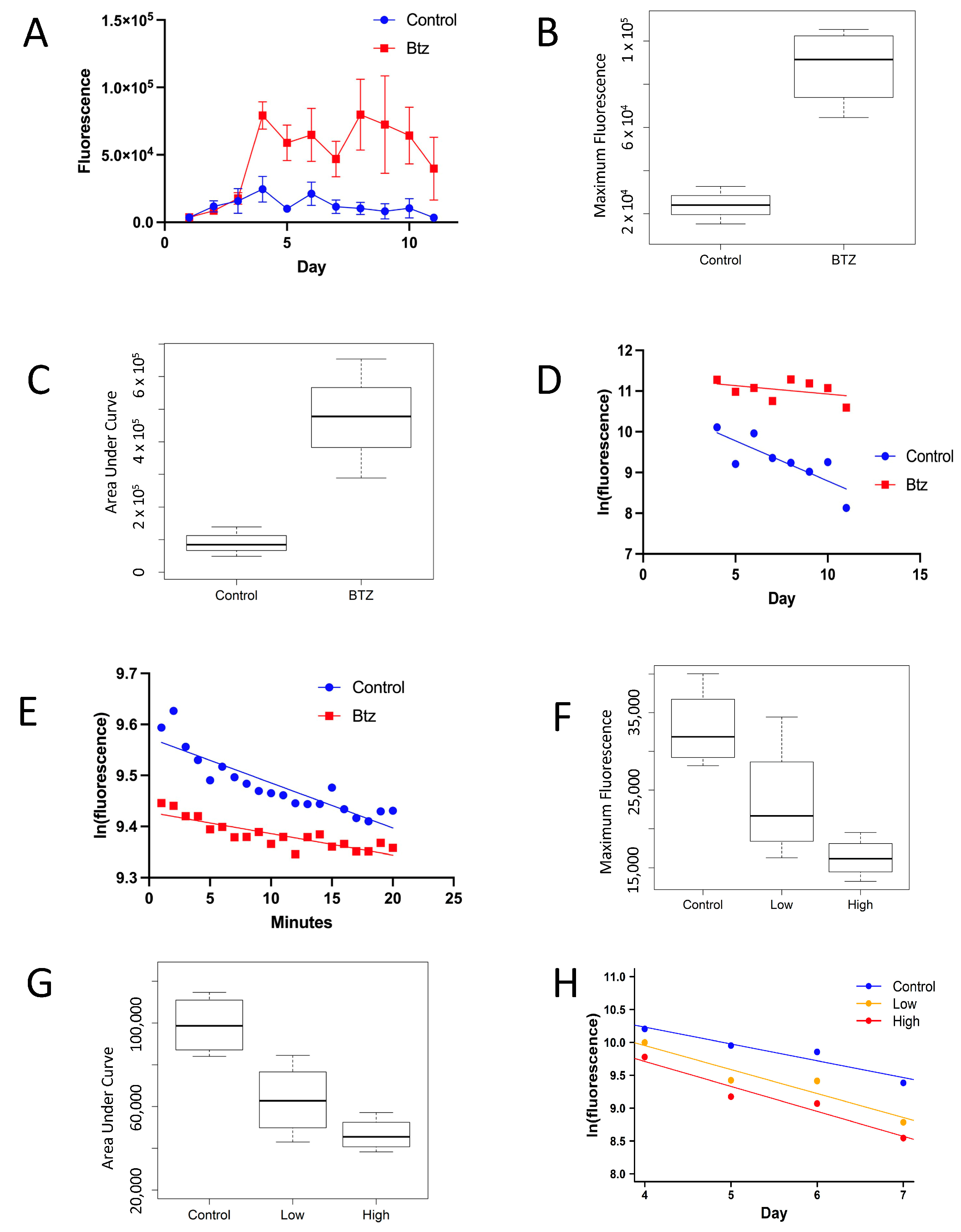
2.6. In Vivo Bortezomib Treatment
Virgin female progeny of Actin-GS-255B x ultraGFP cross were placed in vials with media adjusted to a final concentration of 20 µM bortezomib. The control group media received an equal volume of ethanol vehicle. All flies were also exposed to 200 µg/mL mifepristone in the media for 48 h starting from the first day of videos. Videos were taken every day for 11 days. Flies were switched to fresh ± drug vials every 48 h through the end of the experiment.
2.7. In Vivo Cycloheximide Treatment
Virgin female progeny of the Actin-GS-255B x ultraGFP cross were treated with cycloheximide added to food vials as an aqueous solution. Flies were treated with cycloheximide beginning 7 days before the start of video recordings, at an initial age of 1–3 days. Flies in the low-concentration group were maintained on media adjusted to a final concentration of 5 µM cycloheximide, and the high-concentration group was maintained on media adjusted to a final concentration of 10 µM cycloheximide. Flies in the control group were maintained on vials adjusted with an equal volume of water vehicle. Beginning with the initial video recording, all flies were transferred to vials adjusted to 200 µg/mL mifepristone, in addition to any cycloheximide. Videos were taken every day for 7 days. After 48 h, flies were removed from mifepristone and transferred to new ± cycloheximide vials; flies continued to be switched to fresh drug vials every 48 h through the end of the experiment.
2.8. Microscope Image Capture Assay and Statistical Analysis
Flies with eGFP expression were maintained on NutriFly Grape Agar Premix (Genesee Scientific 47-102), adjusted to 0.5% Casein protein (Sigma/Aldrich). Flies with red fluorescent protein expression were maintained in regular fly media vials. Flies were transferred to fresh vials every other day without anesthetization. For image capture, three flies at a time were anesthetized with CO
2 on a fly pad (Genesee Scientific), positioned on their side, and fluorescence images were generated using the Leica MZFLIII fluorescence microscope and Spot imaging system. Fly fluorescence was quantified using Image J version 1.54 software [
20] and selecting the desired fly body regions as ROI using the free-hand drawing tool. For each time point, >10 flies per sample were quantified, and the data average ± standard deviation was calculated. The ln(fluorescence) versus time was plotted and analyzed using Prism 9 to conduct linear regression and generate half-life values.
2.9. In Vitro Proteasome Assay
In vitro degradation of protein by the proteasome was assayed essentially as previously described [
10]. Young (3–4 day old) virgin female flies from the eyeless-GFP strain were used to generate extracts containing abundant eGFP. Whole flies were homogenized using a motorized pestle in an extraction buffer composed of 50 mM Tris, 25 mM KCl, 10 mM NaCl, 1 mM MgCl
2, and 1 mM DTT at pH 7.5. Three groups of 20 flies each were homogenized in 500 µL of buffer. After homogenization, each sample was subjected to 3 rounds of a freeze/thaw cycle consisting of 5 min in dry ice followed by 5 min in a room-temperature water bath. After 3 cycles the samples were centrifuged for 2 min at 10,000×
g in a 4 °C cold room. The supernatants from the 3 replicates were pooled and then diluted 1:4 in the buffer solution. Next, 180 µL aliquots of the diluted extract were added to black 96-well plates (Greiner Bio-One 82050-728, Kremsmünster, Austria), and 20 µL of either pure ethanol (control) or 250 µM Bortezomib (experimental group) was added to each sample. Purified recombinant eGFP (Cell Biolabs STA-201, San Diego, CA, USA) at 1 mg/mL in PBS was diluted 1:5, 1:10 and 1:15 with PBS, and 200 µL of each sample was loaded into the plate in duplicate as controls. This plate was then introduced to the plate reader pre-warmed to 37 °C. GFP fluorescence was quantified using the BioTek Synergy H4 Hybrid Multi-Mode Microplate Reader (Agilent, Santa Clara, CA, USA) in the USC Dornsife NanoBiophysics Core facility (Los Angeles, CA, USA). Readings were taken at 30 s intervals at 395 nm excitation/510 nm emission to detect GFP fluorescence.
2.10. Generation of Partially Purified eGFP Extract
Young virgin female flies of the eyeless-GFP strain were anesthetized with CO2 on the fly pad, and the heads were removed using a razor blade. A total of 80 heads were combined with 1 mL PBS, homogenized and subjected to 3 rounds of freeze/thaw, centrifuged as described above, and the supernatant used as extract. The extract was diluted 1:2, 1:4, 1:8, and 1:16 in PBS and added to the plate at 180 µL per well. Three replicates of each concentration were supplemented with 20 µL of 250 µM bortezomib, and the controls were supplemented with 20 µL of ethanol vehicle. Reads were conducted using a plate reader as described above, at room temperature (23.5 ± 0.5 °C). GFP fluorescence was quantified using a BioTek Synergy H4 Hybrid Multi-Mode Microplate Reader. For all experiments, readings were taken at 30 s intervals at 395 nm excitation/510 nm emission to detect GFP fluorescence.
2.11. Video Analysis Software and Statistical Analysis
Videos were generated and analyzed essentially as previously described [
9]. The software SaveImageToAviEx_v142.exe is used to record videos at 30 frames per second. The desired number of frames to capture is input by the user, and the resulting video from each camera is recorded and saved as .avi video files. The FluoreScore suite is next used to process these videos into quantitative data. The software FluoreScoreGUI is used to analyze representative videos, select a fluorescence threshold for fly detection, select the ROI, and create a mask if needed to omit any areas with background fluorescence from the analysis. Next, the software template.bat file is used and can be edited for each experiment to indicate the chosen threshold for fluorescence detection, the coordinates for the ROI, and which mask files to utilize, if any. The template.bat file is the only file the user edits and runs directly. It calls the software FluoreScoreCMDV2.exe, which analyzes the videos and derives fluorescence values for the flies in each frame of video, and calls the software SqueezeData_V1.1.exe, which combines the data from the two cameras and filters out signal below the input threshold. The final output is .csv files with total fly fluorescence for each frame of video. The software Cymito_v5.R uses the data from the .csv files to create plots of average fluorescence versus time for each group in the experiment. This software also generates a summary .csv file, with the data in an easy-to-read format, including columns for the average daily fluorescence for every vial, average across all vials, and standard deviation. The data are then plotted as average ln(fluorescence) versus time using Prism 9 to conduct linear regression and generate half-life values. Linear models were also constructed using values from the replicate vials, rather than a single average for each time point, but demonstrated no advantage in identifying differences between groups or constructing lines with a good fit. To conduct ANOVA, the half-life values along with information about the age and sex of the flies were organized into a table, which was then read into R. The R lm function was used to construct a linear model relating half-life to age and sex (including interaction), and then the ANOVA function was used to run the statistical test. Both functions are available in base R version 4.4.
4. Discussion
Here, methods for measuring the half-life of fluorescent proteins in living flies are presented, which enable measurements of protein turnover under different conditions and in distinct tissues and subcellular locations. This assay captures changes in both protein degradation and synthesis. The half-life of eGFP was increased by the proteasome inhibitor bortezomib, both in vivo and in vitro, indicating proteasomal degradation of eGFP. Consistent with the conclusion that eGFP is degraded by the proteasome, bortezomib has previously been shown to inhibit GFP degradation in cultured human H1299 carcinoma cells [
25], and proteasome inhibitors MG132 and Z-L3-VS inhibited GFP degradation in cultured HeLa cells [
26]. It was observed that in the plate reader assay, absolute eGFP fluorescence was slightly lower in the presence of bortezomib. The absolute magnitude of this effect was approximately equal across different concentrations of eGFP, with a larger percent change at lower concentrations (
Supplementary Figure S3). This indicates that the observed effect, a decrease in half-life in the presence of bortezomib, was unlikely to be an artifact of this inhibition, because a preferential decrease in eGFP fluorescence at lower concentrations would tend to produce a steeper rate of decay, and the effect observed with bortezomib is in the opposite direction. Thus, the presence of this inhibitory effect should not affect our conclusions. The inhibitory effect was not detected in the video experiments. The accumulation of eGFP in vivo was decreased by the protein synthesis inhibitor cycloheximide, without affecting half-life.
DsRED protein was expressed in a tissue-general pattern using the Tet-ON system and
Actin5C gene promoter and had a generally greater half-life than was observed for tissue-general expression of eGFP using the Gene-Switch system and
Actin5C gene promoter. For example, the average half-life of eGFP across the video assay experiments was 4.1 days, whereas the average half-life of DsRED across the video assay experiments was 7.8 days (
Supplementary Tables S1, S2 and S4). Consistent with these observations, a previous study of cultured
Drosophila S2 cells reported a half-life for eGFP of approximately 3.8 days and a half-life for DsRED of approximately 8 days [
27]. The similarity between the half-life values previously obtained using cultured
Drosophila S2 cells and the half-life values obtained here for in vivo
Drosophila assays supports the accuracy of our method for measuring in vivo fluorescent protein half-life, as well as its ability to discern differences in the decay rate between different proteins. Limited degradation of mCherry was detected, and examples of decay rates for eGFP, DsRED and mCherry in young virgin females are plotted from peak to minimum on the same
X-axis scale for comparison (
Supplementary Figure S10).
Differences in the decay rates for fluorescent proteins can be exploited for various uses in research. For example, the Repressible Dual Differential Marker (ReDDM) method for lineage tracing takes advantage of the longer half-life of H2B-RFP (red fluorescent protein) relative to mCD8-GFP (green fluorescent protein) to distinguish stem cells from their differentiated progeny [
28]. The half-life of DsRED increased rapidly between eclosion and day 12 of age, with statistically significant increases detected for each of virgin females, mated females, and virgin males. In the future, it may be of interest to ask if the half-life of DsRED continues to increase at greater ages.
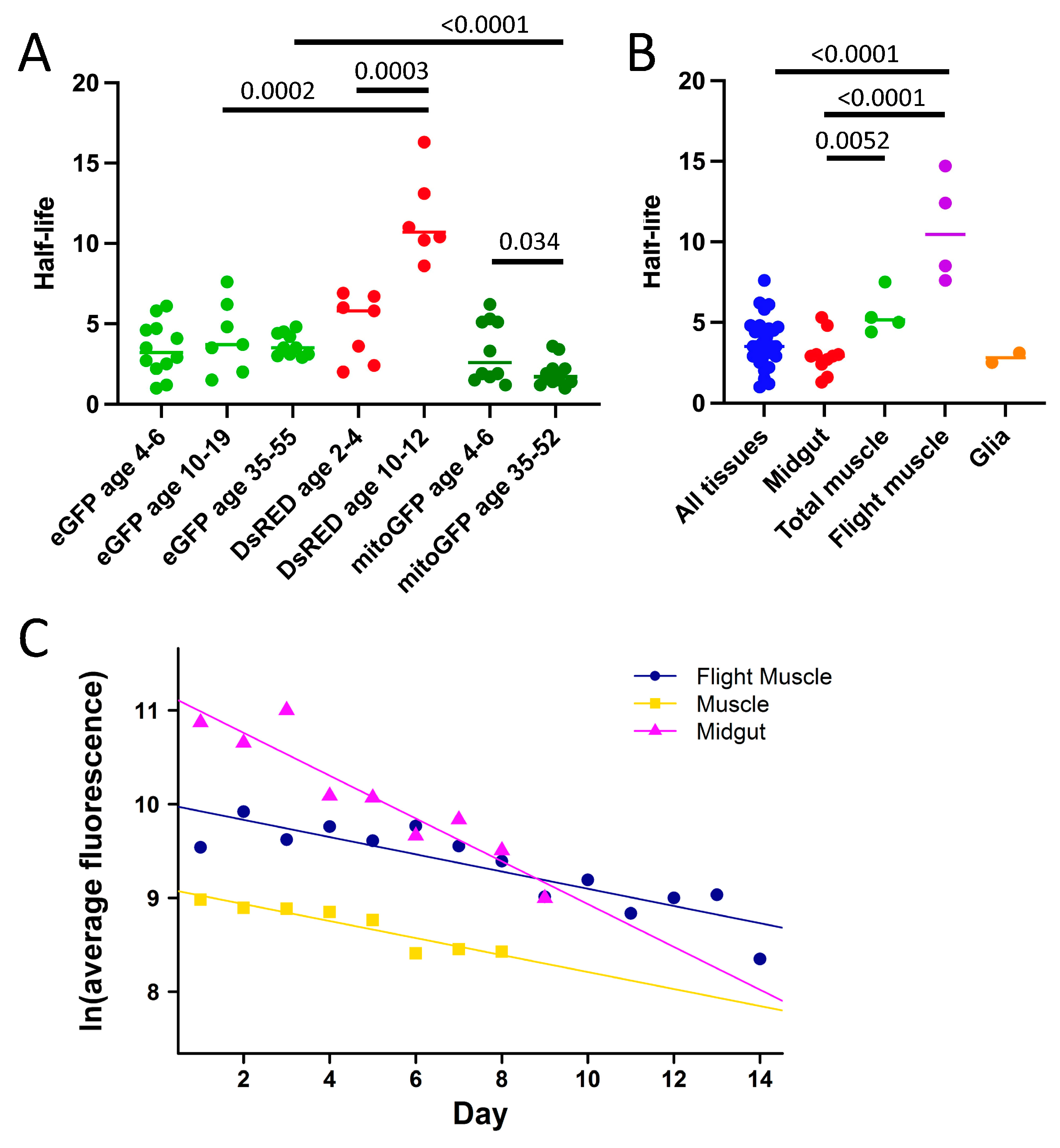
Increased transgene copy number resulted in proportionally increased levels of fluorescent protein expression, as expected (for example, see
Figure S5). This increased fluorescent protein expression is not expected to affect fluorescent protein stability. This is supported by several experiments in which similar half-life values were calculated for the same fluorescent protein despite differences in the maximum fluorescence value (for example, see
Figure 2B). There was no indication of any saturation of the video camera assays or fluorescence microscope assays, such as fluorescence curves that are truncated at the top (for examples, see
Supplemental Figure S8), indicating that the assays are within the linear range. Whereas differences in light penetration might yield somewhat lower fluorescence intensity from internal tissues, we were able to reproducibly assay fluorescent protein half-life in internal tissues such as the gut and nervous system (see
Tables S2 and S5). In the future, increasing the input light intensity might further increase the ability to assay subsets of internal cells.
Half-life also varied by tissue, with greater eGFP half-life observed in muscle relative to other tissues. Cytoplasmic eGFP had a similar half-life in young flies when expressed in a tissue-general pattern and when targeted to midgut enterocytes and to glial cells (summarized in
Figure 3B). In contrast, eGFP half-life was longer in young flies when targeted to muscle tissue (
Figure 3B). The relatively longer half-life of eGFP in
Drosophila muscle tissue may be related to observations that muscle tissue shows preferential accumulation of protein aggregates and induction of hsp70 protein relative to other adult tissues during aging [
29,
30]. These results appear similar to the mouse, where muscle tissue was reported to exhibit generally slower protein turnover than other tissues such as liver [
23]. Limited effects were observed for
Drosophila sex and female mating status. Taken together, the data indicate the in vivo assays are promising tools for the study of protein degradation regulated by protein sequence, subcellular compartment, tissue and small molecules.
Limitations of this study include some asymmetry of the experiments. For example, eGFP has been assayed only using the Gene-Switch system, and DsRED has been assayed only using the Tet-ON system. One reason for this is the relative lack of reagents for the Tet-ON system. Successful expression of eGFP with Tet-ON has been reported in the past, but the tetO-GFP strains are no longer extant [
17]. For both the Gene-Switch and Tet-ON systems, the protein half-life is calculated from the peak of expression to the subsequent minimum. Because the peak in protein expression occurs >24 h after the cessation of drug treatment, it seems unlikely that the differences in the measured half-lives of eGFP and DsRED are a consequence of using different systems but rather are due to inherent differences in the half-lives of the two proteins. In the future, it may be of interest to generate additional fluorescent protein target constructs for the Tet-ON system to allow for direct comparison of half-life values between different proteins without the possible confounding effects of different transgenic systems.
A limitation of the Gene-Switch system is that the mifepristone used to trigger fluorescent protein expression can have significant physiological effects. In mated females, mifepristone reduces progeny production and midgut size and greatly increases median life span [
31]. Limiting mifepristone treatment to 48 h is expected to limit confounding effects. However, it is possible the effects of even brief mifepristone treatment are one reason that differences in eGFP half-life between virgin and mated females were not detected using Gene-Switch. Other conditional gene expression systems each have their own limitations. For example, using heat shock gene promoters and a heat pulse is problematic due to the effects of heat on
Drosophila. In particular, increased temperature reduces the physiological changes associated with mating in female flies, including eliminating the effect of mating on lifespan and midgut size, making heat shock induction unsuitable for comparisons of virgin and mated females [
31]. Moreover, increased temperature shortens lifespan in
Drosophila and can affect protein turnover, altering both synthesis and degradation [
32,
33]. The DOX (doxycycline) drug used to trigger the Tet-ON system has been found to have small effects on mitochondrial translation and gene expression [
34]. Similarly, the Q-system for conditional gene expression is confounded by the fact that the triggering drug, quinic acid, has pleiotropic effects on aging and life span [
35,
36]. Therefore, the use of any conditional gene expression system requires careful consideration of the possible side effects of the system activator.
Additional limitations of the video assay include some variability in results, particularly for the youngest flies. Reasons for this likely include some unequal lighting throughout the vial and some unequal coverage of all regions by the video cameras, combined with fly movement, which is greater in young flies. In future experiments, this variability might be reduced by adding a third camera to improve vial coverage, adding additional lighting, and/or by lengthening video recording time to lessen the impact of fly movement.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}