

Genetic Diversity and Population Structure of Anopheles funestus in Western Kenya Based on Mitochondrial DNA Marker COII
 , , , , ,
, , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mosquito Sampling
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Data Analysis
3. Results
3.1. Genetic Diversity of An. funestus in Western Kenya
3.2. Population Structure and Gene Flow
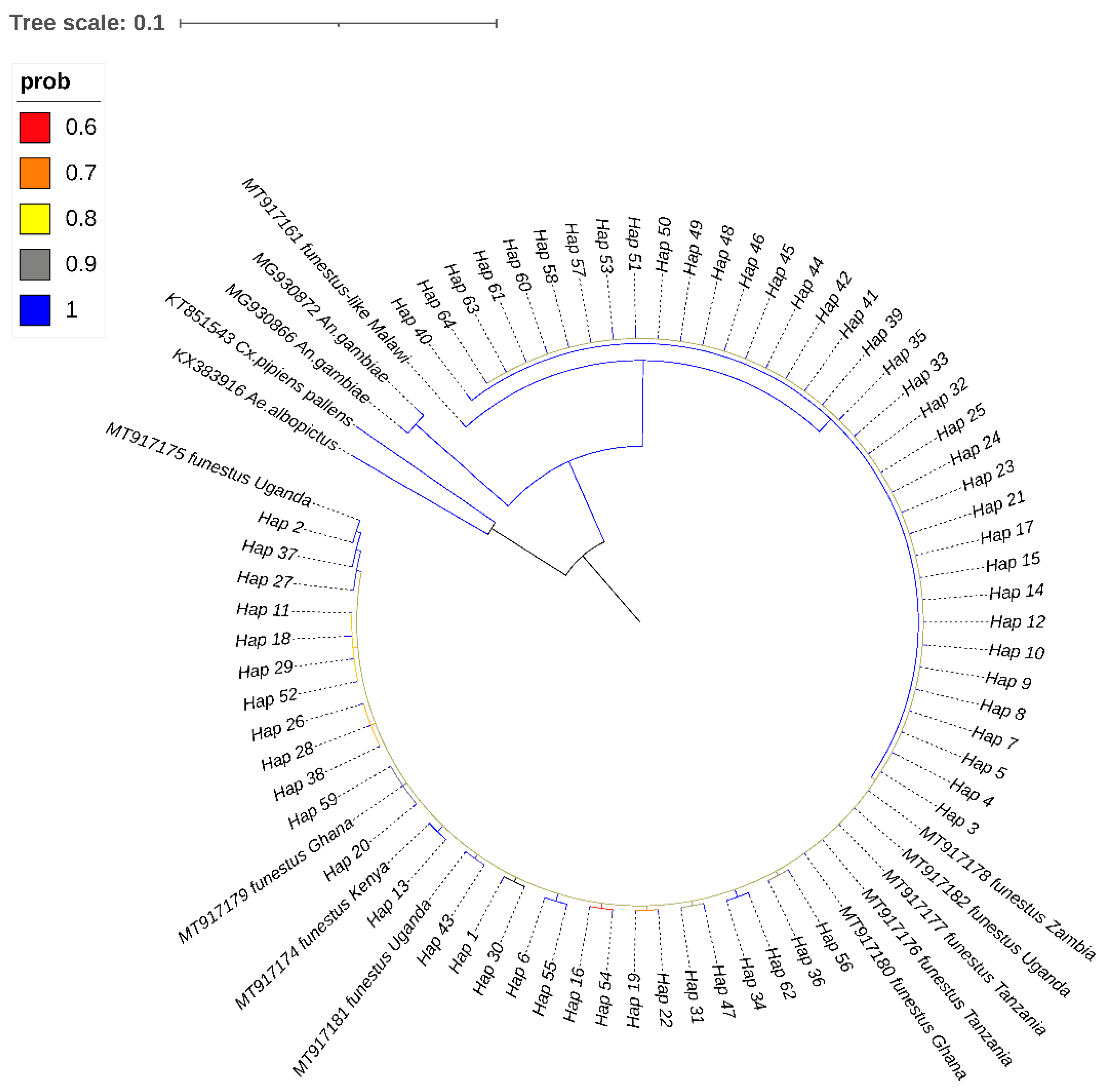
3.3. Phylogenetic Relationships and Network Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sinka, M.E.; Bangs, M.J.; Manguin, S.; Coetzee, M.; Mbogo, C.M.; Hemingway, J.; Patil, A.P.; Temperley, W.H.; Gething, P.W.; Kabaria, C.W. The Dominant Anopheles Vectors of Human Malaria in Africa, Europe and the Middle East: Occurrence Data, Distribution Maps and Bionomic Précis. Parasit. Vectors 2010, 3, 117. [Google Scholar] [CrossRef] [PubMed]
- Harbach, R.E. Mosquito Taxonomic Inventory. 2013. Available online: http://Mosquito-Taxonomic-Inventory.Info/ (accessed on 10 June 2020).
- Garros, C.; Harbach, R.E.; Manguin, S. Morphological Assessment and Molecular Phylogenetics of the Funestus and Minimus Groups of Anopheles (Cellia). J. Med. Entomol. 2005, 42, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Kamau, L.; Munyekenye, G.O.; Koekemoer, L.L.; Hunt, R.H.; Coetzee, M. A Survey of the Anopheles Funestus (Diptera: Culicidae) Group of Mosquitoes from 10 Sites in Kenya with Special Emphasis on Population Genetic Structure Based on Chromosomal Inversion Karyotypes. J. Med. Entomol. 2003, 40, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Makhawi, A.M.; Aboud, M.A.; Raba’a, F.M.E.; Osman, O.F.; Elnaiem, D.E.A. Identification of Anopheles Species of the Funestus Group and Their Role in Malaria Transmission in Sudan. J. Appl. Indust. Sci. 2015, 3, 58–62. [Google Scholar]
- Awolola, T.S.; Oyewole, I.O.; Koekemoer, L.L.; Coetzee, M. Identification of Three Members of the Anopheles Funestus (Diptera: Culicidae) Group and Their Role in Malaria Transmission in Two Ecological Zones in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 2005, 99, 525–531. [Google Scholar] [CrossRef]
- Temu, E.A.; Minjas, J.N.; Tuno, N.; Kawada, H.; Takagi, M. Identification of Four Members of the Anopheles Funestus (Diptera: Culicidae) Group and Their Role in Plasmodium Falciparum Transmission in Bagamoyo Coastal Tanzania. Acta Trop. 2007, 102, 119–125. [Google Scholar] [CrossRef]
- Kaindoa, E.W.; Matowo, N.S.; Ngowo, H.S.; Mkandawile, G.; Mmbando, A.; Finda, M.; Okumu, F.O. Interventions That Effectively Target Anopheles Funestus Mosquitoes Could Significantly Improve Control of Persistent Malaria Transmission in South-Eastern Tanzania. PLoS ONE 2017, 12, e0177807. [Google Scholar] [CrossRef]
- Sougoufara, S.; Diédhiou, S.M.; Doucouré, S.; Diagne, N.; Sembène, P.M.; Harry, M.; Trape, J.-F.; Sokhna, C.; Ndiath, M.O. Biting by Anopheles Funestus in Broad Daylight after Use of Long-Lasting Insecticidal Nets: A New Challenge to Malaria Elimination. Malar. J. 2014, 13, 125. [Google Scholar] [CrossRef]
- Coetzee, M.; Fontenille, D. Advances in the Study of Anopheles Funestus, a Major Vector of Malaria in Africa. Insect Biochem. Mol. Biol. 2004, 34, 599–605. [Google Scholar] [CrossRef]
- Debrah, I.; Afrane, Y.A.; Amoah, L.E.; Ochwedo, K.O.; Mukabana, W.R.; Zhong, D.; Zhou, G.; Lee, M.-C.; Onyango, S.A.; Magomere, E.O.; et al. Larval Ecology and Bionomics of Anopheles Funestus in Highland and Lowland Sites in Western Kenya. PLoS ONE 2021, 16, e0255321. [Google Scholar] [CrossRef]
- Zouache, K.; Fontaine, A.; Vega-Rua, A.; Mousson, L.; Thiberge, J.-M.; Lourenco-De-Oliveira, R.; Caro, V.; Lambrechts, L.; Failloux, A.-B. Three-Way Interactions between Mosquito Population, Viral Strain and Temperature Underlying Chikungunya Virus Transmission Potential. Proc. R. Soc. B Biol. Sci. 2014, 281, 20141078. [Google Scholar] [CrossRef]
- Ogola, E.O.; Odero, J.O.; Mwangangi, J.M.; Masiga, D.K.; Tchouassi, D.P. Population Genetics of Anopheles Funestus, the African Malaria Vector, Kenya. Parasit. Vectors 2019, 12, 15. [Google Scholar] [CrossRef]
- Norris, D.E. Genetic Markers for Study of the Anopheline Vectors of Human Malaria. Int. J. Parasitol. 2002, 32, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Bunmee, K.; Thaenkham, U.; Saralamba, N.; Ponlawat, A.; Zhong, D.; Cui, L.; Sattabongkot, J.; Sriwichai, P. Population Genetic Structure of the Malaria Vector Anopheles Minimus in Thailand Based on Mitochondrial DNA Markers. Parasit. Vectors 2021, 14, 496. [Google Scholar] [CrossRef] [PubMed]
- Weeraratne, T.C.; Surendran, S.N.; Walton, C.; Karunaratne, S.H.P. Genetic Diversity and Population Structure of Malaria Vector Mosquitoes Anopheles Subpictus, Anopheles Peditaeniatus, and Anopheles Vagus in Five Districts of Sri Lanka. Malar. J. 2018, 17, 271. [Google Scholar] [CrossRef]
- Chang, X.; Zhong, D.; Lo, E.; Fang, Q.; Bonizzoni, M.; Wang, X.; Lee, M.-C.; Zhou, G.; Zhu, G.; Qin, Q. Landscape Genetic Structure and Evolutionary Genetics of Insecticide Resistance Gene Mutations in Anopheles Sinensis. Parasit. Vectors 2016, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, C.; Yang, R.; Luo, C.; Deng, Y.; Liu, Y.; Zhou, H.; Zhang, D. Molecular Phylogeny of Anopheles Nivipes Based on MtDNA-COII and Mosquito Diversity in Cambodia-Laos Border. Malar. J. 2022, 21, 91. [Google Scholar] [CrossRef]
- Coetzee, M. Key to the Females of Afrotropical Anopheles Mosquitoes (Diptera: Culicidae). Malar. J. 2020, 19, 70. [Google Scholar] [CrossRef]
- Musapa, M.; Kumwenda, T.; Mkulama, M.; Chishimba, S.; Norris, D.E.; Thuma, P.E.; Mharakurwa, S. A Simple Chelex Protocol for DNA Extraction from Anopheles Spp. JoVE J. Vis. Exp. 2013, 71, e3281. [Google Scholar]
- Koekemoer, L.L.; Kamau, L.; Hunt, R.H.; Coetzee, M. A Cocktail Polymerase Chain Reaction Assay to Identify Members of the Anopheles Funestus (Diptera: Culicidae) Group. Am. J. Trop. Med. Hyg. 2002, 66, 804–811. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Matvienko, M. CLC Genomics Workbench. Plant Anim. Genome Sr. Field Appl. Sci. CLC Bio 2015, 1, 1–42. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Zhong, Y.; Tang, Z.; Huang, L.; Wang, D.; Lu, Z. Genetic Diversity of Procambarus Clarkii Populations Based on Mitochondrial DNA and Microsatellite Markers in Different Areas of Guangxi, China. Mitochondrial DNA Part A 2020, 31, 48–56. [Google Scholar] [CrossRef]
- Higuera, A.; Muñoz, M.; López, M.C.; Reyes, P.; Urbano, P.; Villalobos, O.; Ramírez, J.D. Development of a Multilocus Sequence Typing Scheme for Giardia Intestinalis. Genes 2020, 11, 764. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Collins, F.H.; James, A.A. Genetic Modification of Mosquitoes. Sci. Med. 1996, 3, 52–61. [Google Scholar]
- Dia, I.; Guelbeogo, M.W.; Ayala, D. Advances and Perspectives in the Study of the Malaria Mosquito Anopheles Funestus. Anopheles Mosq. New. Insights Malar. Vectors 2013, 10, 55389. [Google Scholar]
- Braginets, O.P.; Minakawa, N.; Mbogo, C.M.; Yan, G. Population Genetic Structure of the African Malaria Mosquito Anopheles Funestus in Kenya. Am. J. Trop. Med. Hyg. 2003, 69, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Afrane, Y.A.; Vardo-Zalik, A.M.; Atieli, H.; Zhong, D.; Wamae, P.; Himeidan, Y.E.; Minakawa, N.; Githeko, A.K.; Yan, G. Changing Patterns of Malaria Epidemiology between 2002 and 2010 in Western Kenya: The Fall and Rise of Malaria. PLoS ONE 2011, 6, e20318. [Google Scholar] [CrossRef]
- Ototo, E.N.; Mbugi, J.P.; Wanjala, C.L.; Zhou, G.; Githeko, A.K.; Yan, G. Surveillance of Malaria Vector Population Density and Biting Behaviour in Western Kenya. Malar. J. 2015, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Minakawa, N.; Beier, J.; Yan, G. Population Genetic Structure of Anopheles Gambiae Mosquitoes on Lake Victoria Islands, West Kenya. Malar. J. 2004, 3, 48. [Google Scholar] [CrossRef]
- Noor, A.M.; Gething, P.W.; Alegana, V.A.; Patil, A.P.; Hay, S.I.; Muchiri, E.; Juma, E.; Snow, R.W. The Risks of Malaria Infection in Kenya in 2009. BMC Infect. Dis. 2009, 9, 180. [Google Scholar] [CrossRef]
- Samb, B.; Dia, I.; Konate, L.; Ayala, D.; Fontenille, D.; Cohuet, A. Population Genetic Structure of the Malaria Vector Anopheles Funestus, in a Recently Re-Colonized Area of the Senegal River Basin and Human-Induced Environmental Changes. Parasit. Vectors 2012, 5, 188. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population Growth Makes Waves in the Distribution of Pairwise Genetic Differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar]
- Meiklejohn, C.D.; Montooth, K.L.; Rand, D.M. Positive and Negative Selection on the Mitochondrial Genome. Trends Genet. 2007, 23, 259–263. [Google Scholar] [CrossRef]
- Pasteur, N.; Raymond, M. Insecticide Resistance Genes in Mosquitoes: Their Mutations, Migration, and Selection in Field Populations. J. Hered. 1996, 87, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Obando, O.A.; Bona, A.C.D.; Duque L, J.E.; Navarro-Silva, M.A. Insecticide Resistance and Genetic Variability in Natural Populations of Aedes (Stegomyia) Aegypti (Diptera: Culicidae) from Colombia. Zool. Curitiba 2015, 32, 14–22. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Populations | N | L | S | Π | h | Hd | Tajima’s | Fs | Fu and Li’s | Fu and Li’s | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D | p | Statistics | p | D | p | F | p | |||||||
| Port Vic. | 38 | 774 | 32 | 0.005 | 30 | 0.98 | −1.86 | <0.05 | −31.51 | 0.000 | −1.99 | >0.10 | −2.31 | >0.05 |
| Migori | 38 | 774 | 26 | 0.004 | 25 | 0.97 | −1.76 | >0.05 | −21.85 | 0.000 | −2.75 | <0.05 | −2.86 | <0.05 |
| Bungoma | 22 | 774 | 19 | 0.004 | 18 | 0.98 | −1.64 | >0.05 | −16.42 | 0.000 | −1.89 | >0.10 | −2.12 | >0.05 |
| Kombewa | 28 | 774 | 17 | 0.004 | 23 | 0.98 | −1.33 | >0.10 | −25.04 | 0.000 | −1.06 | >0.10 | −1.34 | >0.10 |
| All populations | 126 | 41 | 0.004 | 64 | 0.97 | −1.81 | <0.05 | −32.91 | 0.000 | −1.42 | >0.10 | −1.89 | >0.05 | |
| Populations | No. of Shared Mutations (%) | Dxy | Hs | Ks | Kxy | Gst | Gamma St | Fst | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Value | Nm | Value | Nm | p-Value | |||||||
| Port Vic. vs. Migori | 20/40 (50) | 0.004 | 0.97 | 3.400 | 3.365 | 0.001 | 0.008 | 30.25 | −0.010 | −24.45 | 0.855 |
| Bungoma vs. Kombewa | 12/25 (48) | 0.004 | 0.98 | 2.878 | 2.836 | −0.006 | 0.013 | 19.43 | −0.016 | −16.04 | 0.892 |
| Port Vic vs. Bungoma | 18/34 (52.9) | 0.004 | 0.98 | 3.389 | 3.222 | −0.004 | 0.007 | 35.22 | −0.020 | −12.80 | 0.982 |
| Migori vs. Bungoma | 14/32 (43.8) | 0.004 | 0.97 | 3.048 | 2.994 | 0.001 | 0.013 | 19.02 | −0.008 | −31.71 | 0.649 |
| Port Vic vs. Kombewa | 15/36 (41.7) | 0.004 | 0.98 | 3.324 | 3.234 | −0.003 | 0.011 | 23.43 | −0.009 | −28.29 | 0.838 |
| Migori vs. Kombewa | 15/30 (50) | 0.004 | 0.97 | 3.015 | 2.985 | 0.0003 | 0.014 | 17.99 | −0.003 | −82.77 | 0.468 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debrah, I.; Ochwedo, K.O.; Otambo, W.O.; Machani, M.G.; Magomere, E.O.; Onyango, S.A.; Zhong, D.; Amoah, L.E.; Githeko, A.K.; Afrane, Y.A.; et al. Genetic Diversity and Population Structure of Anopheles funestus in Western Kenya Based on Mitochondrial DNA Marker COII. Insects 2023, 14, 273. https://doi.org/10.3390/insects14030273
Debrah I, Ochwedo KO, Otambo WO, Machani MG, Magomere EO, Onyango SA, Zhong D, Amoah LE, Githeko AK, Afrane YA, et al. Genetic Diversity and Population Structure of Anopheles funestus in Western Kenya Based on Mitochondrial DNA Marker COII. Insects. 2023; 14(3):273. https://doi.org/10.3390/insects14030273
Chicago/Turabian StyleDebrah, Isaiah, Kevin O. Ochwedo, Wilfred O. Otambo, Maxwell G. Machani, Edwin O. Magomere, Shirley A. Onyango, Daibin Zhong, Linda E. Amoah, Andrew K. Githeko, Yaw A. Afrane, and et al. 2023. "Genetic Diversity and Population Structure of Anopheles funestus in Western Kenya Based on Mitochondrial DNA Marker COII" Insects 14, no. 3: 273. https://doi.org/10.3390/insects14030273
APA StyleDebrah, I., Ochwedo, K. O., Otambo, W. O., Machani, M. G., Magomere, E. O., Onyango, S. A., Zhong, D., Amoah, L. E., Githeko, A. K., Afrane, Y. A., & Yan, G. (2023). Genetic Diversity and Population Structure of Anopheles funestus in Western Kenya Based on Mitochondrial DNA Marker COII. Insects, 14(3), 273. https://doi.org/10.3390/insects14030273