Simple Summary
Whiteflies (Bemisia tabaci) ‘B mitotype’ represent an insect pest of fruit, vegetable, ornamental and weed plants. B. tabaci is a cryptic species comprising at least eight endemic haplotypes, of which haplotypes 6 and/or 8 are recognized invasives. The objective of this study was to identify the prevalence and relationships among putative native and exotic begomoviruses and North Africa–Middle East haplotypes in Oman. Several begomoviral species were identified from B. tabaci adults collected from infested plant species, with 67% and 33% representing native and exotic species, respectively. Logistic regression and correspondence analyses predicted ‘strong’ and ‘close’ virus–vector associations involving haplotypes 5 and 2 and the exotic chili leaf curl virus and endemic tomato yellow leaf curl virus-OM, respectively.
Abstract
Irrigated agriculture and global trade expansion have facilitated diversification and spread of begomoviruses (Geminiviridae), transmitted by the Bemisia tabaci (Gennadius) cryptic species. Oman is situated on major crossroads between Africa and South Asia, where endemic/native and introduced/exotic begomoviruses occur in agroecosystems. The B. tabaci ‘B mitotype’ belongs to the North Africa–Middle East (NAFME) cryptic species, comprising at least eight endemic haplotypes, of which haplotypes 6 and/or 8 are recognized invasives. Prevalence and associations among native and exotic begomoviruses and NAFME haplotypes in Oman were investigated. Nine begomoviral species were identified from B. tabaci infesting crop or wild plant species, with 67% and 33% representing native and exotic species, respectively. Haplotypes 2, 3, and 5 represented 31%, 3%, and 66% of the B. tabaci population, respectively. Logistic regression and correspondence analyses predicted ‘strong’- and ‘close’ virus–vector associations involving haplotypes 5 and 2 and the exotic chili leaf curl virus (ChiLCV) and endemic tomato yellow leaf curl virus-OM, respectively. Patterns favor a hypothesis of relaxed virus–vector specificity between an endemic haplotype and the introduced ChiLCV, whereas the endemic co-evolved TYLCV-OM and haplotype 2 virus–vector relationship was reinforced. Thus, in Oman, at least one native haplotype can facilitate the spread of endemic and introduced begomoviruses.
1. Introduction
In Oman, members of the genus Begomovirus (family, Geminiviridae) have been identified as infecting diverse crops, including vegetables, tropical fruit trees, and wild or uncultivated plant species [1]. The begomoviral genome is ~2.7–2.8 kb in size and consists of a circular single-stranded DNA (ssDNA) encapsulated in a particle, having a twinned, quasi-icosahedral or geminate morphology. Begomoviruses have either a monopartite or bipartite genome, DNA-A, or DNA-A and DNA-B components, respectively. Monopartite begomoviruses occur almost exclusively in the Old World (OW) or Eastern Hemisphere, whereas those extant in the New World (NW) have a bipartite genome, except for the NW monopartite begomovirus tomato leaf curl deformation virus (ToLDeV), found to infect tomato plants in Peru [2].
The whitefly Bemisia tabaci Gennadius (Hemiptera: Aleyrodidae) cryptic species group [3,4,5], particularly the B mitotype (or NAFME putative cryptic species), causes damage to crops as a pest and as an insect vector of the genus Begomovirus. In Oman, where it is not native, it colonizes wild plant species and agricultural crops, including beans, cucumber, eggplant, mint, okra, pepper, squash, tobacco, tomato, and watermelon, among others. Begomoviruses are transmitted by B. tabaci in a circulative, non-propagative manner [3]. Studies have shown that the extent of transmission specificity and, subsequently, transmission efficiency or ‘competency’ may be most optimal among phylogeographically congruent begomoviral species–whitefly cryptic species combinations [6,7,8,9]. However, several studies have suggested that certain B. tabaci cryptic or specific haplotypes may be promiscuous in that they may be capable of transmitting one or more begomoviruses with which they have not co-evolved [3,6,10,11,12]. Thus, predicting compatible and incompatible whitefly vector–begomovirus combinations is difficult, despite the overall increased knowledge about whitefly–virion protein–protein interactions that govern viral transmission and of the phylo-biogeographical distribution and microclimate niche affiliations of whitefly–begomovirus combinations, which may collectively define boundaries or give rise to determinants of specificity (phenotypes) [13].
Differentiation of B. tabaci populations at a finer level, such as by microsatellites mitotypes (mtCOI) [3] and haplotypes (mtCOI SNPs), have been used to resolve information on co-evolutionary lineages and associated begomoviral populations, strains, and species. In a recent study, the B. tabaci mtCOI sequence was shown to be evolving 12.5 times faster than the nuclear genome [4], making it a valuable marker for discerning micro-evolutionary scale changes. The availability of the mtCOI sequence for a large sample size of the B mitotype/cryptic species has facilitated the identification of eight SNPs haplotypes of the ‘B mitotype’ or NAFME cryptic species, NAFME 1–8, which have either strict or overlapping geographical affinities as well as microclimate niches within. The NAFME 6 and 8 are now recognizable as the pervasive, exotic B. tabaci haplotypes that have invaded locales in Asia, Africa, Australia, Europe, the Mediterranean region, and the New World [14]. Haplotypes NAFME 1–3 co-exist in the desert regions of the Arabian Peninsula. The prototype NAFME 3 field isolate was first identified in Ethiopia in the 1990s. The sister haplotypes NAFME 4 and 5 are predominant in the semi-arid niches of Iran and Pakistan. Haplotypes NAFME 6–8 appear to be adapted to both the Mediterranean and desert climates found in Israel and Egypt, leading to the hypothesis that NAFME 6 dispersed/spread eastward to Asia, while NAFME 8 spread westward and colonized the New World and elsewhere [14]. Finally, in a previous study, haplotype NAFME 5 was identified in Nizwa, Oman, thus far the only location where it has been documented [14]. Additional sampling is expected to reveal whether NAFME 1–3 haplotypes may also occur here, which would be consistent with macroclimate predictions of the NAFME 1–3 endemism.
The B mitotype and its haplotypes [14] and many previously described monopartite begomoviruses are endemic to the area encompassing northeastern Africa (adjacent to the Red Sea), the Arabian Peninsula, and southwestern Asia [3]. Transmission studies with China-native or recently introduced B mitotypes and Cotton leaf curl Multan virus (CLCuMuV-Fai), introduced from the Indian subcontinent to China, have shown that CLCuMuV-Fai is transmissible by a laboratory-maintained colony of an Asia II 1 mitotype, which is native to parts of both China and the Indian subcontinent. However, colonies of a China-native Asia 1 mitotype and of the non-native B and Q mitotypes prevalent in agroecosystems in China did not transmit the introduced CLCuMuV-Fai isolate [15,16,17,18,19]. These and other reports of less-than-strict begomovirus-vector specificity between endemic–non-endemic virus–vector combinations [6,12,20,21] have challenged the paradigm that B. tabaci–begomovirus relationships do not necessarily coincide with co-endemism, as has been previously hypothesized [13]. Thus, understanding the extent to which begomovirus-B. tabaci mitotype and/or haplotype transmission specificity has evolved, with a strict or relaxed basis in co-endemism, could greatly inform the risk assessment of exotic and non-exotic virus–vector combinations with small- or global-scale invasion potential.
The objective of this study is to determine whether the distribution of exotic (introduced) and native whitefly B. tabaci haplotypes in Oman are associated with extant native or introduced begomoviral species. Plants exhibiting symptoms reminiscent of begomovirus infection and B. tabaci whiteflies infesting symptomatic plants were collected from different governorates in Oman during the growing seasons and analyzed for begomovirus presence. Begomoviruses were provisionally identified by sequence analysis of a 688-base pair (bp) fragment of the begomoviral cp. Identification of whitefly B. tabaci haplotypes was based on informative mtCOI SNPs previously shown to differentiate the eight known haplotypes associated with the NAFME cryptic species [14]. A correspondence analysis (CA) and logistic regression were carried out to identify potential associations between the occurrence of whitefly haplotype and begomoviral species.
2. Material and Methods
2.1. Whitefly and Plant Field Collections
Adult whiteflies were collected from whitefly-infested crop plants and weed species during 2015–2018 from 34 sites in seven Oman Governorates. Multiple samples were collected from symptomatic cultivated or uncultivated (wild) host plants exhibiting leaf curling, vein thickening, and/or enations, indicative of virus-like diseases. Five to ten whitefly adults were collected from infested plants, approximately ~500 feet apart, per collection site. Each whitefly sample consisted of a minimum of five adults infesting the leaves of an infested plant. Whiteflies were collected live using a hand-held aspirator, transferred to a 1.5-mL microfuge tube containing 95% alcohol, and stored at −20 °C (Table 1). For each sample, the collector’s name, date of collection, and field location GPS coordinates were recorded (Table 1). For plant samples, leaf samples were collected from the growing tip of symptomatic vegetable, ornamental, or wild (uncultivated) plants (n = 36), exhibiting begomovirus-like symptoms consisting of leaf curling, mosaic, or mottling, shortened internodes, and overall stunting. Leaf samples collected from each symptomatic plant were placed in a separate plastic bag, transferred to an insulated container with ice, transported to the laboratory, and stored at −4 °C (Table 1).

Table 1.
Sample number, year of collection, location of collection with GPS coordinates, host plant, agroecosystem, provisional begomoviral species associated with plant and whitefly samples, begomoviral predicted center(s) of diversification, GenBank Accession number for begomoviral and mitochondrial cytochrome oxidase I (COI) sequence, and Bemisia tabaci haplotype based on SNPs analysis.
2.2. Total DNA Isolation from Plants and Whiteflies, PCR Amplification, DNA Sequencing, and Sequence Analysis
Total nucleic acids were isolated from leaves collected from symptomatic plant samples and from individual whiteflies using the E.Z.N.A.® Insect DNA Kit (Omega Bio-tek, Norcross, GA, USA) for plant tissue and whiteflies, respectively, a commercial kit based on the CTAB method [22].
2.3. Begomoviral Coat Protein Gene Fragment
The begomoviral coat protein fragment was amplified from total genomic DNA. A 688-base pair (bp) fragment of the begomoviral species-informative region of the coat protein (‘core cp’) was obtained by PCR amplification from DNA purified from the plant using the primers AV1-F/R (5′-ATCATTTCCACKCCCGYCTCGA-3′/5′-GCRTGMGTACABGCCATATACA-3′) [23]. The AV1 fragment is nested within the coat protein region (cp) of the begomovirus and has been validated extensively as a phylogenetically informative sequence for tentative begomovirus species identification. Cycling parameters were denaturation at 93 °C for 30 s, followed by 30 cycles of 93 °C for 30 s, 55 °C for 30 s, 72 °C for 40 s, and a final extension at 72 °C for 10 min. The PCR product size was estimated by agarose gel (1.5%) electrophoresis (TAE buffer, pH 8.0), and visualized by staining with ethidium bromide (Thermo Fisher Scientific, CA, USA). The PCR amplicons were cloned into the pTZ57R/T plasmid vector (Thermo Fisher Scientific, CA, USA). The plasmids containing an insert of the expected size were confirmed by restriction enzyme analysis and inserts of the expected size were sequenced completely by Sanger DNA sequencing (Macrogen Inc.; Seoul, Republic of Korea).
Preliminary begomovirus identification was carried out by phylogenetic analyses and by subjecting core cp fragments to a BLASTn [24] search of the NCBI, using a virus reference sequence database to identify the top hits. Sequences representing the top twenty hits with the highest similarity score, at 99–100%, coverage (95–100%), and e-values of less than 10−2 were downloaded from the GenBank database. Downloaded references (accessed on 16 March 2021) were used to build phylogenetic trees. Phylogenetic analysis was carried out using the Bayesian algorithm [25], and the tree was drawn with TreeAnnotator v1.84 [26] and visualized with FigTree v1.42 (http://tree.bio.ed.ac.uk/software/figtree/, accessed on 1 March 2023).
Pairwise distances for the core cp sequences were calculated using the Sequence Demarcation Tools (SDT) software v1. 2 [27], and a cut-off of <88–91% was used to assign the provisional species names [28]. Previous calibrations of core cp region divergence over the last twenty years (authors, unpublished results) indicated that pairwise distance comparisons of the core cp with the respective complete genome sequence were only slightly higher and did not result in discrepancies in species predictions, except those with predicted recombination within in the vicinity of the coat protein and/or the cp as well as leftward or rightward sequences that could be documented. Here, the phylogenetic analysis prediction (provisional core cp) of species placements on the tree was consistent with the results of BLASTn (GenBank virus Refseq) and pairwise distances (SDT) analyses.
2.4. Whitefly Mitochondria Cytochrome Oxidase I Fragment
Each adult whitefly was ground individually with a micropipette-melted tip and homogenized in 350 μL CTL Buffer and 25 μL Proteinase K Solution (50–100 µg/mL) in a 1.5 mL microcentrifuge tube, followed by incubation at 60 °C for 30 min. An equal volume of chloroform:isoamyl alcohol (24:1) was added, and the solution was vortexed and centrifuged (Eppendorf 5452 Mini Spin Centrifuge) at 10,000× g for 3 min. The supernatant was mixed with one volume of BL Buffer (Omega Bio-tek, Inc., Norcross, GA, USA) and 2 μL RNase A, followed by incubation at 70 °C for 10 min. The total DNA preparation was column-purified using a HiBind® DNA Mini Column (Omega Bio-tek, Inc., Norcross, GA, USA). The HiBind® DNA Mini Column was washed with HBC and DNA wash buffers according to the manufacturer’s instructions. The total purified DNA was eluted in 10 mM-Tris-EDTA buffer and stored at −20 °C.
The 3′-end fragment of the mtCOI gene was PCR-amplified using primers (C1-J-2195 5′–TTGATTTTTTGGTCATCCAGAAGT–3′; TL2-N-3014 5′–TCCAATGCACTAATCTGCCATATTA–3′) [29] to obtain an expected size product of 865 bp. Cycling conditions were as follows: denaturation at 93 °C for 30 s, followed by 35 cycles of 93 °C for 30 s, 52 °C for 30 s, 72 °C for 45 s, and a final extension at 72 °C for 10 min. At least two amplicons per sample of the expected size were cloned into the pTZ57R/T plasmid vector (Thermo Fisher Scientific, California, USA). Plasmids containing the expected size of the insert were confirmed by restriction digestion. The confirmed cloned inserts were sequenced (Sanger) bi-directionally (Macrogen Inc.; Seoul, Republic of Korea).
2.5. SNPs Classification of Haplotypes of North African–Middle East B Mitotype
The mtCOI sequences were screened to identify informative single nucleotide polymorphisms (SNPs) [14] in Geneious Prime v2021.1.1 using the ‘Find Variants’ tool, using 2% as the minimum variant frequency. The Arizona B biotype prototype (GenBank accession AY057123) was included as the reference sequence. The SNP profiles were compared to a recently published global alignment of NAFME haplotypes [14] that groups the NAFME (B) haplotypes into eight NAFME clades by phylogenetic analysis using a Maximum Likelihood (ML) algorithm. The RaxML phylogenetic tree was constructed using 100 bootstrap iterations, optimized substitution rates, and the GAMMA model of rate heterogeneity. The B. tabaci putative MEAM2, which is recognized as a pseudogene (GenBank accession MT605827), was used to root the tree.
2.6. Spatial Distribution of Bemisia Tabaci NAFME Haplotypes
The geographic distribution map of the NAFME haplotypes identified within Oman and reference sequences determined for other locations in the Middle East were drawn using the geographic coordinates for each sample. Maps of administrative divisions were downloaded from the online geographic information systems platform DIVA-GIS (https://www.diva-gis.org/, accessed on 1 March 2023), and occurrence records of NAFME haplotypes were compiled in QGIS v3.8.1 (QGIS development team, 2021) using the ‘Biological Records Tool’ plugin.
2.7. Correlation and Correspondence Analyses of Whitefly Vector–Begomovirus Associations
The significance of associations between whitefly haplotypes and begomovirus species detected in whitefly and plant tissues were tested using a logistic regression approach using the ‘glm’ function of the ‘stats’ package v4.2.0 (R Core Team 2021). The PCR amplification results were entered into a text matrix as a binomial variable, where 1 and 0 corresponded to the presence and absence of a begomoviral species, respectively. The begomovirus species identified in adult whiteflies and plant samples were evaluated to determine if potentially significant correlations existed between the occurrence and identity of the whitefly vector haplotype, except when a begomoviral species was detected in a single plant or whitefly sample. The correspondence analysis was carried out using the software InfoStat v.2020 [30] by tabulating a contingency table, with Euclidean distances assigned to each pair of categories within each discrete variable under study. Each pair of categories was drawn in a biplot (two-dimensional plot) on two axes coordinates that define the Euclidean distance between them [31]. The variance explained by one of two axes of the biplot was given by the ‘eigenvalues’, represented as the proportion of the contribution of each axis that explained the total variability of the data.
3. Results
3.1. Classification of Whitefly Haplotypes Based on SNPs and Phylogeographical Relationships
The 34 mtCOI sequences determined in this study were differentiated as haplotypes based on one to eight SNPs identified in the 3′-end of the COI gene [14]. The AY057123 GenBank sequence was used as the reference mtCOI sequence for SNP identification. The AY057123 sequence corresponds to the prototype AZ-B (previously, ‘biotype’) (Brown Lab, UA Tucson, Arizona), recognized as an invasive B. tabaci introduced in the US in approximately 1988–1989 [3]. The predominant SNP profile corresponded to haplotype NAFME 5 and revealed four nucleotide transitions, one thiamine, and three cytosine residues at positions 826, 1137, 1152, and 1375, respectively. NAFME 2 was characterized by having four fixed cytosine residues at positions 810, 1068, 1137, and 1152, respectively, a transition from a guanine to an adenine residue at position 1053, and a transversion from a thymine to a guanine residue at position 1074. The NAFME 3 haplotype shared fixed nucleotides with NAFME 2 at positions 1068, 1074, 1137, and 1152 of the mtCOI gene [16] (Supplementary Figure S1). No whiteflies analyzed from the Oman collections belonged to two recently recognized invasive haplotypes, NAFME 6 or 8, of which only NAFME 8 has a near-cosmopolitan distribution on several continents. Similarly, the phylogenetic (ML) analysis resolved the whitefly haplotypes from Oman into one of the three NAFME clades 2, 3, and 5 in relation to previously published reference sequences (Figure 1 and Supplementary Figure S1) [14].
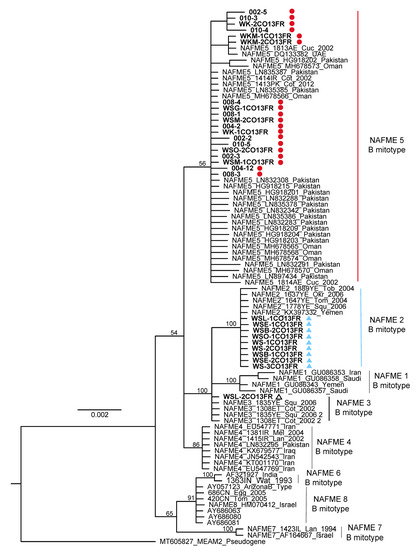
Figure 1.
Maximum likelihood tree for mitochondrial cytochrome oxidase I gene sequences of the NAFME haplotype groups, reconstructed using RaxML. Whitefly haplotypes NAFME 2, 3, and 5 grouped most closely with reference sequences belonging to the North Africa–Middle East (NAFME) clade. One thousand bootstrap iterations were carried out, and statistical support values (>60%) were placed at the major nodes.
The NAFME 2 and 5 haplotype distributions overlap geographically, with both inhabiting the northern and southern regions of Oman. However, NAFME 5 was by far the most abundant haplotype, followed by NAFME 2, at 66 and 31%, respectively. Among the B. tabaci whiteflies collected in this study, only the collection from Al-Batinah North was identified as NAFME 3 (Figure 1 and Figure 2).
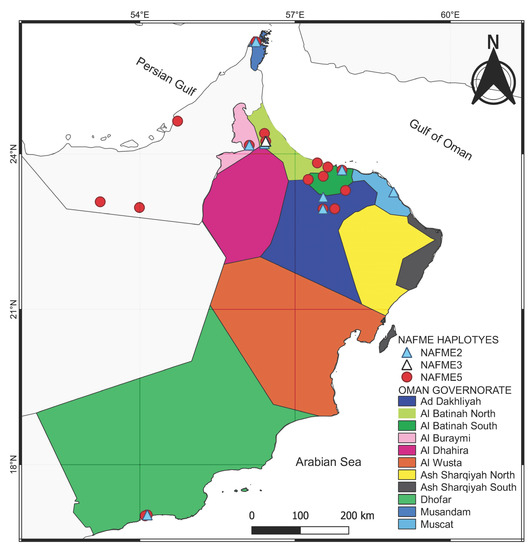
Figure 2.
The distribution of the Bemisia tabaci belonging to the North Africa–Middle East (NAFME) cryptic species in Oman.
3.2. Provisional Begomovirus Identification and Species Associated with NAFME Haplotypes
Of thirty-six collected whitefly samples, sixteen confirm the presence of nine different begomovirus species ((Supplementary Figure S2 and Table 1). These begomoviruses, considered to be native or to have originated from Oman and surrounding locales, were Tomato leaf curl Barka virus (ToLCBrV), Tomato leaf curl Liwa virus (ToLCLwV), Tomato yellow leaf curl virus-Oman (TYLCV-OM), and Watermelon chlorotic stunt virus (WmCSV), while the exotic viruses known to be introduced from Africa are Cotton leaf curl Gezira virus (CLCuGeV) and Tomato leaf curl Sudan virus (ToLCSDV) from Asia, with Chili leaf curl virus (ChiLCV), Mungbean yellow mosaic virus (MYMIV), and Squash leaf curl virus (SLCV) having their origins in the Americas (New World). We detected ChiLCV, ToLCBrV, ToLCLwV, and TYLCV-OM begomovirus species from whitefly haplotypes. Among these, the begomovirus detection rate was highest for ChiLCV (50%), followed by TYLCV-OM (37.5%), whereas ToLCBrV and ToLCLwV were each detected at a frequency of 6.25%.
NAFME 2 showed the largest rate of virus detection at 66.7%, followed by 42.1% for NAFME 5 (Figure 3). Unexpectedly, begomoviral amplicons were not detected for the B. tabaci NAFME 3 haplotype. The detection rate of ChiLCV for NAFME 5 and NAFME 2 was 31.6% and 11%, respectively. The detection rate of TYLCV-OM was 44.4% and 5% for NAFME 2 and NAFME 5, respectively. Additionally, ToLCLwV and ToLCBrV were associated with the NAFME 2 haplotype at an 11% detection rate each (Figure 3). The geographic distribution of ChiLCV and TYLCV-OM was found to be widespread; however, they varied in prevalence and by location within the five Governorates of Al-Batinah, Al-Musandam, Dhofar, Ad-Dakhilya, and Ash Sarkyah. In contrast, the begomoviral species, CLCuGeV, MYMIV, ToLCBrV, ToLCLwV, ToLCSDV, and WmCSCV, were detected in B. tabaci-infested host plants in the mainland Al-Batinah Governorate only and were associated with either NAFME 5 and NAFME 2 haplotypes at low detection rates of 11% or below.
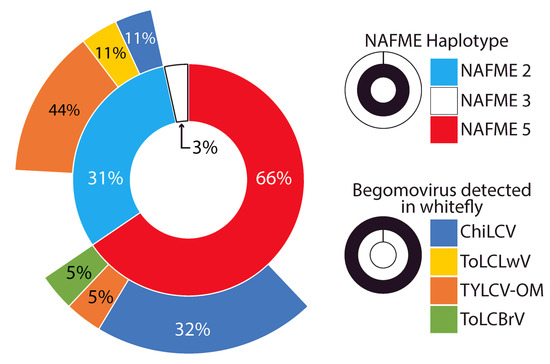
Figure 3.
Frequency of Bemisia tabaci NAFME haplotypes (inner chart layer) and begomoviral species (outer chart layer) detected in whitefly extracts.
B. tabaci whiteflies were collected from host species from different agroecosystems, including vegetables grown in greenhouses and home gardens, field crops, fruit crops, and wild, uncultivated plant species (Table 1). The NAFME 2 haplotype was the predominant B. tabaci in the Dhofar Governorate, which is located about 1000 km from the main agriculture area in Al-Batinah. Haplotype 2 was associated with begomovirus-infected cucurbit species, mint, and tomato plants (Table 1). The NAFME 5 haplotype occurred throughout Oman and colonized begomovirus-infected papaya, pepper, tobacco, tomato, watermelon, and wild (uncultivated) host species. The NAFME 3 haplotype was detected in the Musandam governorate, where it occurred on pumpkin plants, a species from which ChiLCV has been detected in other collection sites, albeit not in this location (Table 1).
Nine begomovirus species were provisionally identified based on PCR amplification of the core coat protein gene from B. tabaci adults. Most of the begomoviruses detected were associated with B. tabaci-infested vegetable crop species, including cucumber, mint, pepper, pumpkin, tobacco, tomato, and watermelon. Begomoviruses were also identified in papaya trees and several weeds. The non-native ChiLCV was identified in B. tabaci collected from papaya, pepper, mint, tobacco, tomato, and an unidentified species of Urtica. Whiteflies collected from cucumber, papaya, pumpkin, and tomato were shown to harbor the native begomovirus TYLCV-OM, while whiteflies collected from tomato plants were infected with two endemic begomoviruses, ToLCVBrV and ToLCLwV. However, no begomoviral species were amplified by PCR from NAFME 3 whiteflies collected in the Musandam governorate (Table 1).
3.3. Associations between NAFME Haplotypes and Begomoviruses
A linear regression model was implemented using the logistic regression method to assess the significance of putative associations between the whitefly haplotypes and begomovirus (es) detected in plants and whitefly adults. The logistic regression coefficients indicated that the probability of NAFME 5 harboring TYLCV-OM was significantly decreased compared to the probability of NAFME 2 harboring the same begomovirus species (p = 0.0186), indicating that a strong association was supported between TYLCV-OM and the NAFME 2 haplotype. In contrast, the probability of NAFME 2 harboring ChiLCV was significantly decreased compared to the probability of an association with NAFME 5 (p = 0.0499), suggesting a strong correlation between haplotype 5 and ChiLCV prevalence. Correspondence analysis, with 53% of total variance explained, was consistent with the logistic regression test results, predicting a strong association between TYLCV-OM and ChiLCV and NAFME 2 and 5, respectively. For begomoviral species detected/identified in whitefly-infested plants, the correspondence analysis results indicated a positive association (p < 0.05) between NAFME 2 and the begomoviral species ToLCLwV, ToLCSDV, and TYLCV-OM. Similarly, ChiLCV, CLCuGeV, SLCuV, and ToLCBrV were strongly associated (p < 0.05) with the NAFME 5 haplotype collections (Figure 4). Positive associations were not observed (unsupported) for MYMIV and WmCSV and NAFME haplotypes 1–3.
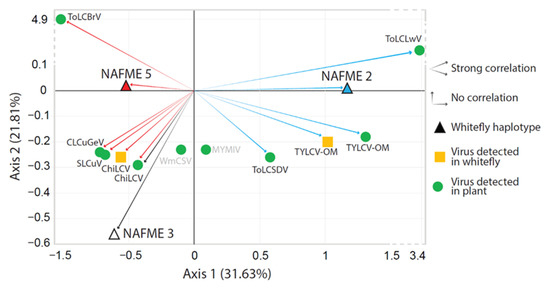
Figure 4.
Correspondence analyses between whitefly haplotypes NAFME 2, 3, and 5 and begomovirus species detected in whitefly and plant samples. Red arrows show positive associations between begomovirus species and whitefly haplotype NAFME 5; blue arrow indicates positive associations between begomovirus species and NAFME 2 haplotype. Black arrow shows the association between the single begomovirus species detected in symptomatic plants infested by the NAFME 3 haplotype.
4. Discussion
4.1. Classification of Whitefly Haplotypes Based on SNPS and Phylogeographical Relationships
The NAFME cryptic species [4], also referred to as the ‘B mitotype’ based on mtCOI typing [3,32] and, previously, as MEAMI cryptic species [33], comprises at least eight phylogeographic haplotype groups, designated as NAFME 1–8 [14]. Only two of these haplotypes, NAFME 6 and 8, have become invasive (e.g., haplotype 8 in the Americas and haplotype(s) 6 and/or 8 in China/Asia), establishing themselves in locales distant from their center of origin in the North Africa–Middle East region (NAFME) [14]. The distribution of the other six haplotypes, their vector competence, and interactions in their native habitats have remained poorly studied until recently. In this study, a country-wide sampling of whiteflies and associated plant tissues was conducted in agroecosystems in Oman to better understand the association between native NAFME haplotypes and the indigenous or exotic begomovirus species they transmit in the desert regions of the Arabian Peninsula, one of three major macro-niches that native NAFME haplotypes inhabit [14].
Based on these results, three NAFME haplotype groups (MEAMI species) were differentiated based on the unique SNPs that correlate to the haplotypes previously identified in the Middle East—NAFME 2, 3, and 5, with NAFME 5 being the predominant haplotype, at a frequency of 66% of the total samples analyzed. Previous studies have shown that NAFME 5 is also the predominant haplotype in Iran and southern Pakistan, suggesting its region of endemism is characterized by the semi-arid climates of the Irano-Turanian region and, potentially, one or more fragmented microclimate niches in the southern deserts of Pakistan [14]. Haplotypes NAFME 2 and 5 are known to occur in the Dhofar Governorate, near the Yemen border in southern Oman, and 1000 km away, in the Al-Batinah, the main agriculture production area in Oman. Additionally, these two NAFME haplotypes have been identified in Khasab in the Al-Musandam Governorate, northwards and near the border of the UAE, 500 and 1500 km from the Al-Batinah and Dhofar regions, respectively. Further, NAFME 2 and 5 overlap in Salalah, where a unique topography is created by the Al-Hajar mountain range that divides the country into northern and southern Oman. The range of NAFME 2 and 5 was found to overlap, despite the low frequency of NAFME 2, at 31%. Additional sampling will be required to determine if the less than 50:50 representation of the two haplotypes might indicate the impending displacement of NAFME 2 by NAFME 5. Such widespread distribution of NAFME 2 and 5 haplotypes in Oman may be the result of ethological variables at play, including but not limited to their flight behavior. In laboratory studies, the B mitotype (now recognized as haplotype NAFME 8) of B. tabaci has been demonstrated to utilize primarily ‘trivial flight’ or engage in relatively short-distance flights between plant hosts; however, trapping studies conducted under field conditions have shown they also exhibit migratory behavior, dispersing long distances on low winds (~20 ft above the landscape), as far as 2.7 km and much further over time [34,35,36,37].
The NAFME 3 haplotype of B. tabaci was identified only in Al-Batinah (north), suggesting either a highly restricted distribution and, potentially, unique microclimate characteristics [38] and/or that future collections or surveys would benefit from a greater sample size to determine the factors that limit the distribution of this B. tabaci haplotype, which is apparently rare in Oman agroecosystems. Previous studies have demonstrated and/or inferred the propensity for phenotypic differences within and among B. tabaci and its cryptic species [3]. Among the recognized phenotypic differences are host range, microclimatic factors, and, among the haplotypes in Oman, potential resource competition between NAFME 2 and 5. The broad and/or limited distribution of B. tabaci mitotypes has been documented in the Indian subcontinent for selected mitotypes in Pakistan, Asia II 1, 7, and 5, and for the A and B mitotypes in the American Tropics. In this study, the two haplotypes documented at the lowest frequency, NAFME 2 and NAFME 3, were collected from vegetation in the urban landscape and household gardens rather than in monoculture cropping systems. This pattern is consistent with observations of native B. tabaci mitotypes in Ecuador and Pakistan, respectively [39]. In this study, the NAFME 5 haplotype colonized a broad range of plant host species/families, which is a previously recognized phenotype of the invasive NAFME 8; for at least some well-studied B. tabaci, albeit in previous historical surveys, the cryptic species of B. tabaci could not yet be ascertained [40].
4.2. Provisional Begomovirus Identification and Species Associated with NAFME Haplotypes
The results of the survey designed to detect begomoviral prevalence in plant host(s) and in the prospective whitefly vector haplotypes of B. tabaci indicated an association of ChiLCV, CLCuGeV, MYMIV, SLCV, ToLCBrV, ToLCLwV, TYLCV-OM, ToLCVSDV, and WmCSV, which were identified from plant hosts, whereas four of them, ChiLCV, ToLCBrV, TYLCV-OM, and ToLCLwV were detected only in whiteflies.
The nine monopartite and/or bipartite begomoviruses identified in this study were found to infect one or more plant hosts, primarily vegetable crops, cucumber, eggplant, mungbean, muskmelon, okra, pepper, pumpkin, tobacco, tomato, and watermelon; two ornamentals–herbs: mint (Lamiaceae) and Senna spp. (Fabaceae); an annual tropical fruit, papaya; and an uncultivated species, Urtica spp. (Urticaceae), collectively (Table 1), representing seven plant families. In Oman, at least fourteen begomoviral species (monopartite and bipartite), including those provisionally identified herein, have been reported previously from vegetable crops, including the monopartite ChiLCV, Chickpea chlorotic dwarf virus (CpCDV), CLCuGeV, Okra leaf curl Oman virus (OLCOMV), ToLCABV, ToLCBrV, ToLCLwV, ToLCOMV, ToLCSDV, the bipartite East African cassava mosaic Zanzibar virus (EACMZV), MYMIV, SLCV, Tomato leaf curl Palampur virus (ToLCPlV), and WmCSV [1,41,42,43,44,45].
4.3. Associations between NAFME Haplotypes and Begomoviruses
Based on logistic regression and correspondence analyses, the various begomovirus-host plant–whitefly haplotype combinations were not equally associated with the two most prominent B. tabaci haplotypes, NAFME 2 and 5. Instead, ChiLCV, of Indian subcontinent endemism, was ‘strongly associated’ (78%) with NAFME 5, a haplotype predicted to be native to Iran and, also possibly, Pakistan, whereas the B. tabaci haplotype NAFME 2 was ‘strongly associated’ (72%) with the type TYLCV-OM strain, extant in Oman.
Among the collection sites in Oman, the NAFME 5 haplotype colonized many species of plants and was associated with as many as nine begomoviral species detected in the plant host or putative whitefly vector (Table 1). These observations predict that B. tabaci haplotype 5 harbors transmission competency for begomovirus species extant in Oman, with which it has and has not co-evolved. Similarly, the NAFME 2 haplotype was predicted to exhibit a strong association with two native (endemic) begomoviral species, TYLCV-OM and ToLCLwV, with which it is known to have co-evolved. The scenario for TYLCV-OM was more complex than for other begomoviruses identified in Oman agroecosystems, primarily because the geographical region (extant), comprising Iran and the Arabian Peninsula, is the recognized ancestral center of diversification of all strains and species of the TYLCV group, of which five are extant in Iran, including TYLCV-OM [44], whereas the TYLCV species group representation in Oman is limited to TYLCV-OM. The genome sequence of several begomoviruses extant in Oman, including the predominant and widespread TYLCV-OM species, exhibit extensive genetic variability, indicating ongoing diversification in Iran and Oman, likely due to geographic isolation. Indeed, the distribution of the haplotype NAFME 5, which is native to a unique microclimate niche occurring in Iran and Pakistan, respectively [14], suggests that NAFME 5 has potentially co-evolved with all the seven recognized TYLCV species and strains [44], albeit, in Oman, the NAFME 5 haplotype was not preferentially strongly associated with TYLCV-OM. Notably, TYLCV and its seven recognized species/strains have not been reported to occur in Pakistan, despite the occurrence of the NAFME 5 haplotype in the deserts of southern Pakistan, located in Bahawalpur and Sindh, respectively, regions of sparse agriculture [21]. However, the potential for the occurrence of TYLCV-OM in south Pakistan cannot be ruled out, given the recent report of the emergence of a new TYLCV-OM species identified from cluster bean (guar) and tomato, Tomato leaf curl Oman virus (ToLCOMV-PK, previously, TYLCV-PK) and its associated tomato leaf curl betasatellite, ToLCB, a predicted recombinant between TYLCV-OM and Papaya leaf curl virus (PaLCV) [45,46], the latter betasatellite, which is considered native to the Indian subcontinent.
The results of the logistic regression and correspondence analysis also predicted a ‘strong association’ between NAFME 5 and exotic/introduced begomoviruses prevalent in plant hosts in Oman (p = 0.0499). The latter introduced viruses, CLCuGeV, MYMIV, SLCV, and WmCSV, are endemic to Asia, the Middle East, Africa, and the Americas, respectively. There is knowledge and evidence of their transmission in Oman by the predicted, non-native NAFME 5 B. tabaci [1,47,48] and in other locations worldwide, where it has been established only recently following its introduction [3]. This scenario does not support the paradigm that B. tabaci cryptic species vector competency and a predicted co-evolution with begomoviral species are linked.
Collectively, these observations suggest that the evolution of vector competence may not be strictly determined by long-standing co-evolutionary forces but rather shorter and near-term adaptation, in line with a set of specific environmental conditions and/or repeated exposures, resulting in promiscuous evolutionary or ‘relaxed’ co-evolutionary outcomes that promote survival and continued diversification. Understanding the basis for begomovirus–whitefly transmission specificity, evolved under strict or relaxed conditions, is paramount to identifying the mechanisms underlying whitefly vector transmission specificity and subsequent competency. This, together with precise environmental and phylo-biogeographical data, importantly, could aid in predicting the begomovirus-B. tabaci cryptic species combinations are most likely to result in locally restricted or long-distance dispersal events, with the potential to lead to outbreaks and/or widespread epidemics.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/insects14030268/s1, Figure S1: SNP profiles; Figure S2: Begomoviruses_sequencesRaXMLtree.
Author Contributions
All authors contributed to the conception and design of the research. Sample collection and archiving and field data collection were carried out by M.S.S., M.A. and A.M.A.-S. Data analyses were carried out by M.S.S. and J.R.P.-M. The first draft, revisions, and final draft of the manuscript were written by M.S.S., J.R.P.-M., M.A. and J.K.B. All authors have read, commented on, and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Sultan Qaboos University under Deanship of Research Grants no. RF/AGR/CROP/19/01 and IG/AGR/CROP/23/02 to M.S.S., and by a grant from Cotton Incorporated, USA (no. 06-829), to J.K.B.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated during this research have been included in this published article.
Conflicts of Interest
Each author has no relevant financial or non-financial interest to disclose.
References
- Khan, A.J.; Mansoor, S.; Briddon, R.W. Oman: A case for a sink of begomoviruses of geographically diverse origins. Trends Plant Sci. 2014, 19, 67–70. [Google Scholar] [CrossRef]
- Melgarejo, T.A.; Kon, T.; Rojas, M.R.; Paz-Carrasco, L.; Zerbini, F.M.; Gilbertson, R.L. Characterization of a new world monopartite begomovirus causing leaf curl disease of tomato in Ecuador and Peru reveals a new direction in geminivirus evolution. J. Virol. 2013, 87, 5397–5413. [Google Scholar] [CrossRef]
- Brown, J.K. Phylogenetic biology of the Bemisia tabaci sibling species group. In Bemisia: Bionomics and Management of a Global Pest; Springer: Berlin/Heidelberg, Germany, 2009; pp. 31–67. [Google Scholar]
- De Moya, R.S.; Brown, J.K.; Sweet, A.D.; Walden, K.K.; Paredes-Montero, J.R.; Waterhouse, R.M.; Johnson, K.P. Nuclear orthologs derived from whole genome sequencing indicate cryptic diversity in the Bemisia tabaci (Insecta: Aleyrodidae) complex of whiteflies. Diversity 2019, 11, 151. [Google Scholar] [CrossRef]
- Hadjistylli, M.; Roderick, G.K.; Brown, J.K. Global population structure of a worldwide pest and virus vector: Genetic diversity and population history of the Bemisia tabaci sibling species group. PLoS ONE 2016, 11, e0165105. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Bird, J.; Frohlich, D.; Rosell, R.; Bedford, I.; Markham, P. The relevance of variability within the Bemisia tabaci species complex to epidemics caused by subgroup III geminiviruses. In Bemisia 1995: Taxonomy, Biology, Damage, Control and Management; Gerling, G., Mayer, R.T., Eds.; Intercept: Andover, MA, USA, 1995; pp. 77–89. [Google Scholar]
- Deng, D.; McGrath, P.; Robinson, D.; Harrison, B. Detection and differentiation of whitefly-transmitted geminiviruses in plants and vector insects by the polymerase chain reaction with degenerate primers. Ann. Appl. Biol. 1994, 125, 327–336. [Google Scholar] [CrossRef]
- Idris, A.M.; Shahid, M.S.; Briddon, R.W.; Khan, A.J.; Zhu, J.K.; Brown, J.K. An unusual alphasatellite associated with monopartite begomoviruses attenuates symptoms and reduces betasatellite accumulation. J. Gen. Virol. 2011, 92 Pt 3, 706–717. [Google Scholar] [CrossRef] [PubMed]
- McGrath, P.F.; Harrison, B.D. Transmission of tomato leaf curl geminiviruses by Bemisia tabaci: Effects of virus isolate and vector biotype. Ann. Appl. Biol. 1995, 126, 307–316. [Google Scholar] [CrossRef]
- Díaz, F.; Endersby, N.M.; Hoffmann, A.A. Genetic structure of the whitefly Bemisia tabaci populations in Colombia following a recent invasion. Insect Sci. 2015, 22, 483–494. [Google Scholar] [CrossRef]
- Gabarra, R.; Alomar, Ò.; Castañé, C.; Goula, M.; Albajes, R. Movement of greenhouse whitefly and its predators between in-and outside of Mediterranean greenhouses. Agric. Ecosyst. Environ. 2004, 102, 341–348. [Google Scholar] [CrossRef]
- Gautam, S.; Mugerwa, H.; Buck, J.W.; Dutta, B.; Coolong, T.; Adkins, S.; Srinivasan, R. Differential Transmission of Old and New World Begomoviruses by Middle East-Asia Minor 1 (MEAM1) and Mediterranean (MED) Cryptic Species of Bemisia tabaci. Viruses 2022, 14, 1104. [Google Scholar] [CrossRef]
- Brown, J.K. The Bemisia tabaci complex: Genetic and phenotypic variation and relevance to TYLCV-vector interactions. In Tomato Yellow Leaf Curl Virus Disease; Springer Nature: Cham, Switzerland, 2007; pp. 25–56. [Google Scholar]
- Paredes-Montero, J.R.; Haq, Q.; Mohamed, A.A.; Brown, J.K. Phylogeographic and SNPs analyses of Bemisia tabaci B mitotype populations reveal only two of eight haplotypes are invasive. Biology 2021, 10, 1048. [Google Scholar] [CrossRef]
- Pan, H.; Chu, D.; Yan, W.; Su, Q.; Liu, B.; Wang, S.; Li, R. Rapid spread of tomato yellow leaf curl virus in China is aided differentially by two invasive whiteflies. PLoS ONE 2012, 7, e34817. [Google Scholar] [CrossRef]
- Pan, L.; Chen, Q.; Guo, T.; Wang, X.; Li, P.; Liu, S. Differential efficiency of a begomovirus to cross the midgut of different species of whiteflies results in variation of virus transmission by the vectors. Sci. China Life Sci. 2018, 61, 1254–1265. [Google Scholar] [CrossRef]
- Ashfaq, M.; Hebert, P.D. DNA barcodes for bio-surveillance: Regulated and economically important arthropod plant pests. Genome 2016, 59, 933–945. [Google Scholar] [CrossRef]
- Ellango, R.; Singh, S.T.; Rana, V.S.; Gayatri Priya, N.; Raina, H.; Chaubey, R.; Asokan, R. Distribution of Bemisia tabaci genetic groups in India. Environ. Entomol. 2015, 44, 1258–1264. [Google Scholar] [CrossRef]
- Islam, W.; Akutse, K.S.; Qasim, M.; Khan, K.A.; Ghramh, H.A.; Idrees, A.; Latif, S. Bemisia tabaci-mediated facilitation in diversity of begomoviruses: Evidence from recent molecular studies. Microb. Pathog. 2018, 123, 162–168. [Google Scholar] [CrossRef]
- Götz, M.; Winter, S. Diversity of Bemisia tabaci in Thailand and Vietnam and indications of species replacement. J. Asia-Pac. Entomol. 2016, 19, 537–543. [Google Scholar] [CrossRef]
- Al-Shihi, A.A.; Hanson, P.; Al-Sadi, A.M.; Al-Yahyai, R.A.; Briddon, R.W.; Deadman, M.; Shahid, M.S. Evaluation of tomato inbred lines for resistance to the tomato yellow leaf curl disease complex in Oman. Crop Prot. 2018, 110, 91–98. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Shahid, M.S.; Raza, A.; Al-Sadi, A.M.; Briddon, R.W. Identification of Chilli leaf curl virus associated with tomato leaf curl betasatellite infecting Mentha in Oman. Can. J. Plant Pathol. 2019, 41, 291–295. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Brown, J.K.; Idris, A.M.; Torres-Jerez, I.; Banks, G.K.; Wyatt, S.D. The core region of the coat protein gene is highly useful for establishing the provisional identification and classification of begomoviruses. Arch. Virol. 2001, 146, 1581–1598. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Di Rienzo, J.; Casanoves, F.; Balzarini, M.; Gonzalez, L.; Tablada, M.; Robledo, C.W. InfoStat Versión 2013, Grupo InfoStat, FCA, Universidad Nacional de Córdoba, Argentina. 2013. Available online: http://www.Infostat.com.ar (accessed on 16 March 2021).
- Greenacre, M.J.; Groenen, P.J. Weighted euclidean biplots. J. Classif. 2016, 33, 442–459. [Google Scholar] [CrossRef]
- Frohlich, D.; Torres-Jerez, I.; Bedford, I.; Markham, P.G.; Brown, J. A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 1999, 8, 1683–1691. [Google Scholar] [CrossRef]
- Saleh, D.; Laarif, A.; Clouet, C.; Gauthier, N. Spatial and host-plant partitioning between coexisting Bemisia tabaci cryptic species in Tunisia. Popul. Ecol. 2012, 54, 261–274. [Google Scholar] [CrossRef]
- Blackmer, J.; Byrne, D. Environmental and physiological factors influencing phototactic flight of Bemisia tabaci. Physiol. Entomol. 1993, 18, 336–342. [Google Scholar] [CrossRef]
- Blackmer, J.; Byrne, D.; Tu, Z. Behavioral, morphological, and physiological traits associated with migratory Bemisia tabaci (Homoptera: Aleyrodidae). J. Insect Behav. 1994, 8, 251–267. [Google Scholar] [CrossRef]
- Byrne, D.N. Migration and dispersal by the sweet potato whitefly, Bemisia tabaci. Agric. For. Meteorol. 1999, 97, 309–316. [Google Scholar] [CrossRef]
- Naranjo, S.E.; Castle, S.J.; Barro, P.J.D.; Liu, S.-S. Population dynamics, demography, dispersal and spread of Bemisia tabaci. In Bemisia: Bionomics and Management of a Global Pest; Springer: Berlin/Heidelberg, Germany, 2009; pp. 185–226. [Google Scholar]
- Paredes-Montero, J.R.; Ibarra, M.A.; Arias-Zambrano, M.; Peralta, E.L.; Brown, J.K. Phylo-biogeographical distribution of whitefly Bemisia tabaci (Insecta: Aleyrodidae) mitotypes in Ecuador. Ecosphere 2020, 11, e03154. [Google Scholar] [CrossRef]
- Cock, M. Bemisia tabaci, an Update 1986–1992 on the Cotton Whitefly with an Annotated Bibliography; CAB IIBC Silwood Park: Berks, UK, 1993. [Google Scholar]
- Akhtar, S.; Khan, A.J.; Singh, A.S.; Briddon, R.W. Identification of a disease complex involving a novel monopartite begomovirus with beta- and alphasatellites associated with okra leaf curl disease in Oman. Arch. Virol. 2014, 159, 1199–1205. [Google Scholar] [CrossRef]
- Shafiq, M.; Sattar, M.N.; Shahid, M.S.; Al-Sadi, A.M.; Briddon, R.W. Interaction of Watermelon chlorotic stunt virus with satellites. Australas. Plant Pathol. 2021, 50, 117–128. [Google Scholar] [CrossRef]
- Shahid, M.S.; Al-Sadi, A.M. First identification of Tomato leaf curl Palampur virus in Oman: Detection and characterization. J. Plant Prot. Res. 2022, 62, 295–301. [Google Scholar]
- Shahid, M.S. Characterization of Tomato leaf curl Palampur virus naturally infecting wild melon in Oman. Indian Phytopathol. 2022, 76, 215–221. [Google Scholar] [CrossRef]
- Hosseinzadeh, M.R.; Shams-Bakhsh, M.; Osaloo, S.K.; Brown, J.K. Phylogenetic relationships, recombination analysis, and genetic variability among diverse variants of Tomato yellow leaf curl virus in Iran and the Arabian Peninsula: Further support for a TYLCV center of diversity. Arch. Virol. 2014, 159, 485–497. [Google Scholar] [CrossRef]
- Zaidi, S.; Shakir, S.; Farooq, M.; Amin, I.; Mansoor, S. First report of a novel strain of Tomato yellow leaf curl virus causing yellow leaf curl disease on cluster bean in Pakistan. Plant Dis. 2017, 101, 1071. [Google Scholar]
- Ahmed, N.; Zaidi, S.S.-E.-A.; Amin, I.; Scheffler, B.E.; Mansoor, S. Tomato leaf curl Oman virus and associated Betasatellite causing leaf curl disease in tomato in Pakistan. Eur. J. Plant Pathol. 2021, 160, 249–257. [Google Scholar] [CrossRef]
- Al Shihi, A.A.; Al Sadi, A.M.; Deadman, M.; Briddon, R.W.; Shahid, M.S. Identification of a distinct strain of Cotton leaf curl Gezira virus infecting tomato in Oman. J. Phytopathol. 2018, 166, 199–205. [Google Scholar] [CrossRef]
- Shahid, M.S. Molecular and biological characterization of Chilli leaf curl virus and associated betasatellite infecting Cucurbita maxima in Oman. Virus Dis. 2020, 31, 378–382. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).