
Precise Characterization of Bombyx mori Fibroin Heavy Chain Gene Using Cpf1-Based Enrichment and Oxford Nanopore Technologies

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genomic DNA Extraction
2.2. PCR
2.3. Cpf1/crRNA Preparation
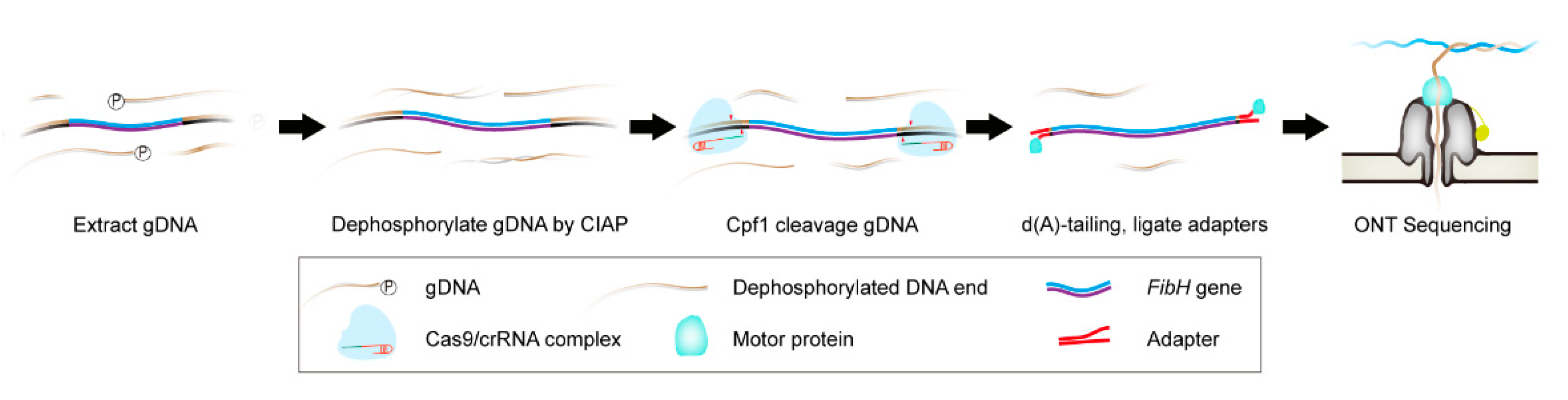
2.4. Nanopore Sequencing Library Preparation
2.5. Data Analysis
3. Results
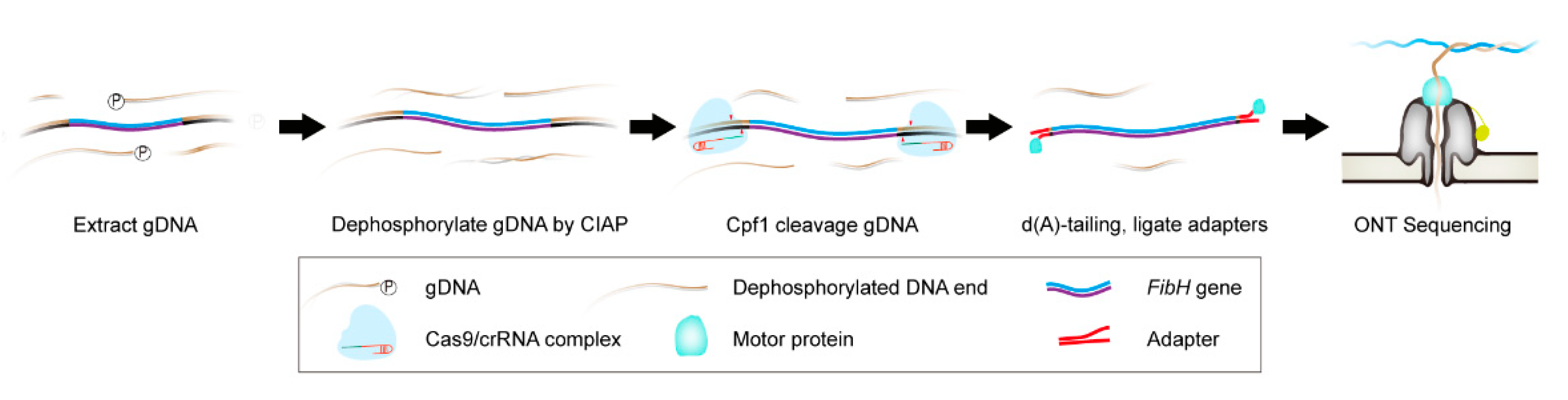
3.1. Cpf1-Based Enrichment and ONT Sequencing (CEO) Overview
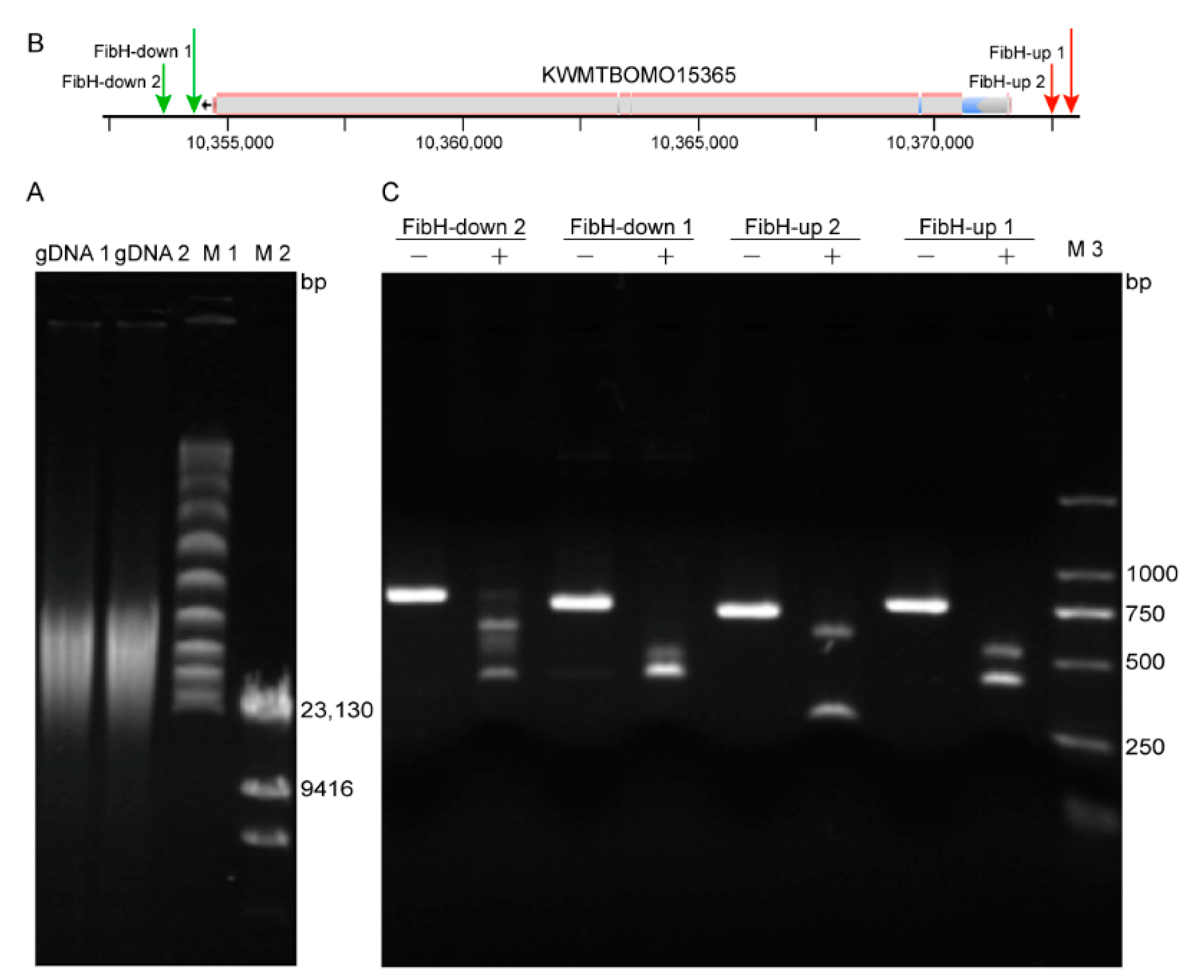
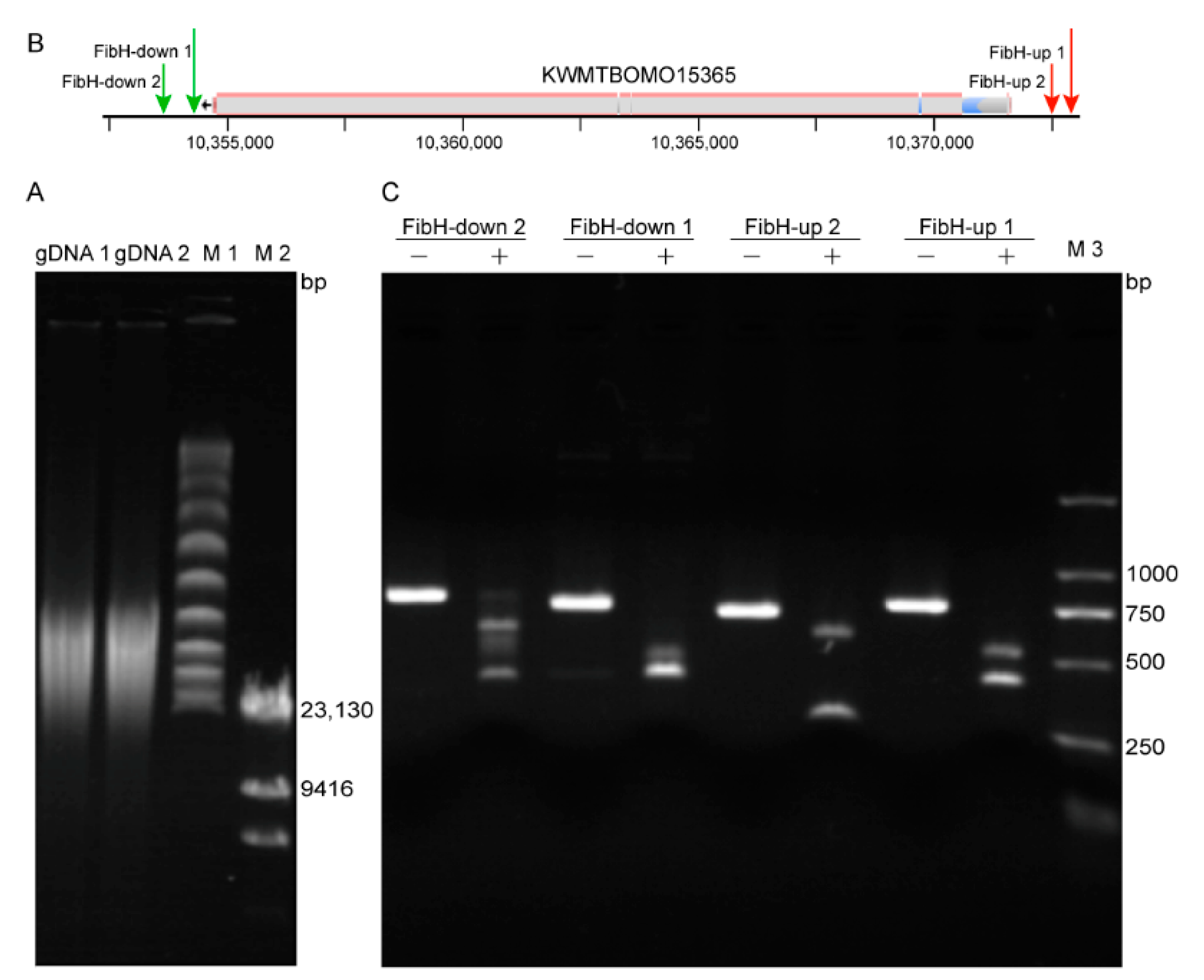
3.2. Preparation of High-Quality B. mori gDNA and High Activity crRNA
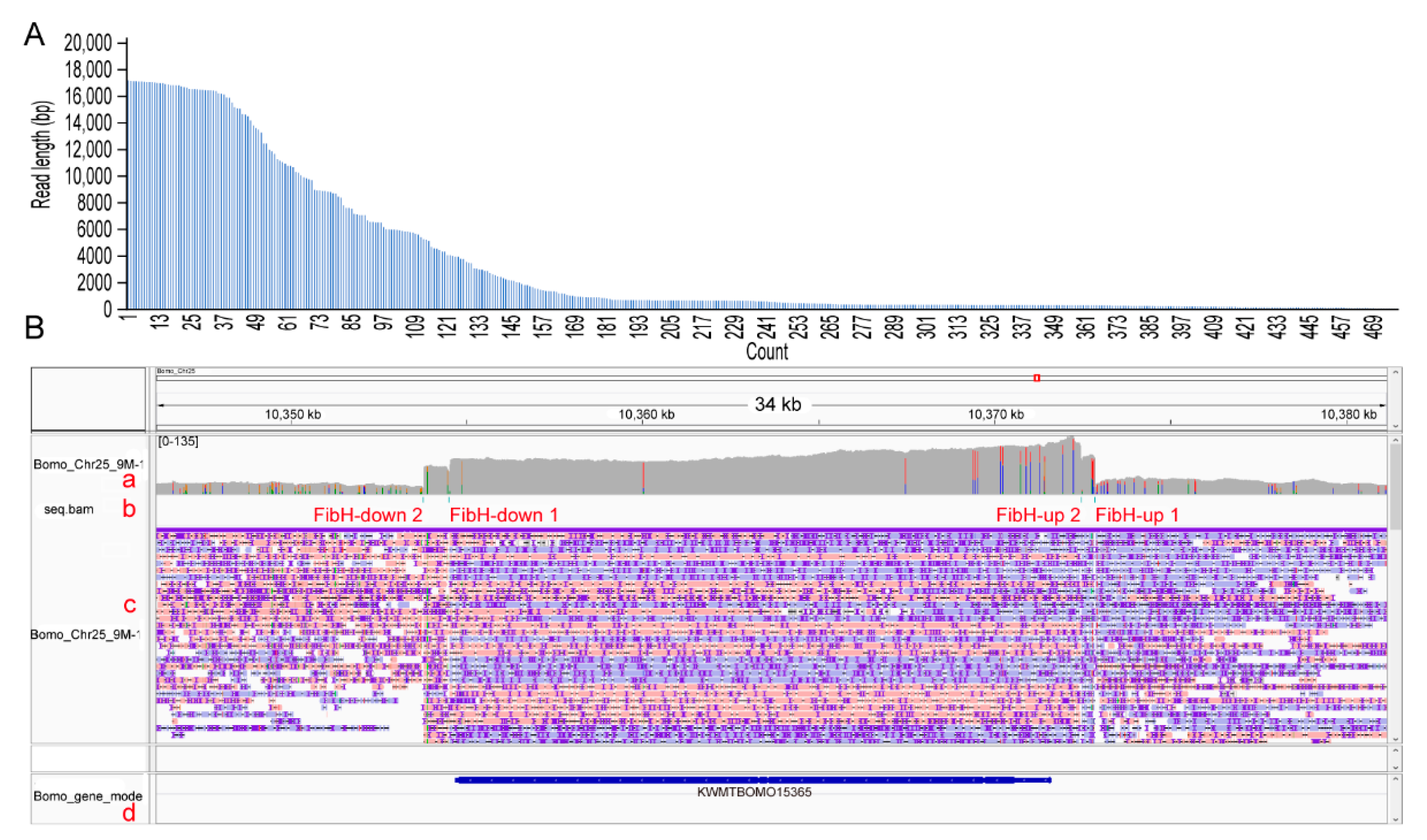
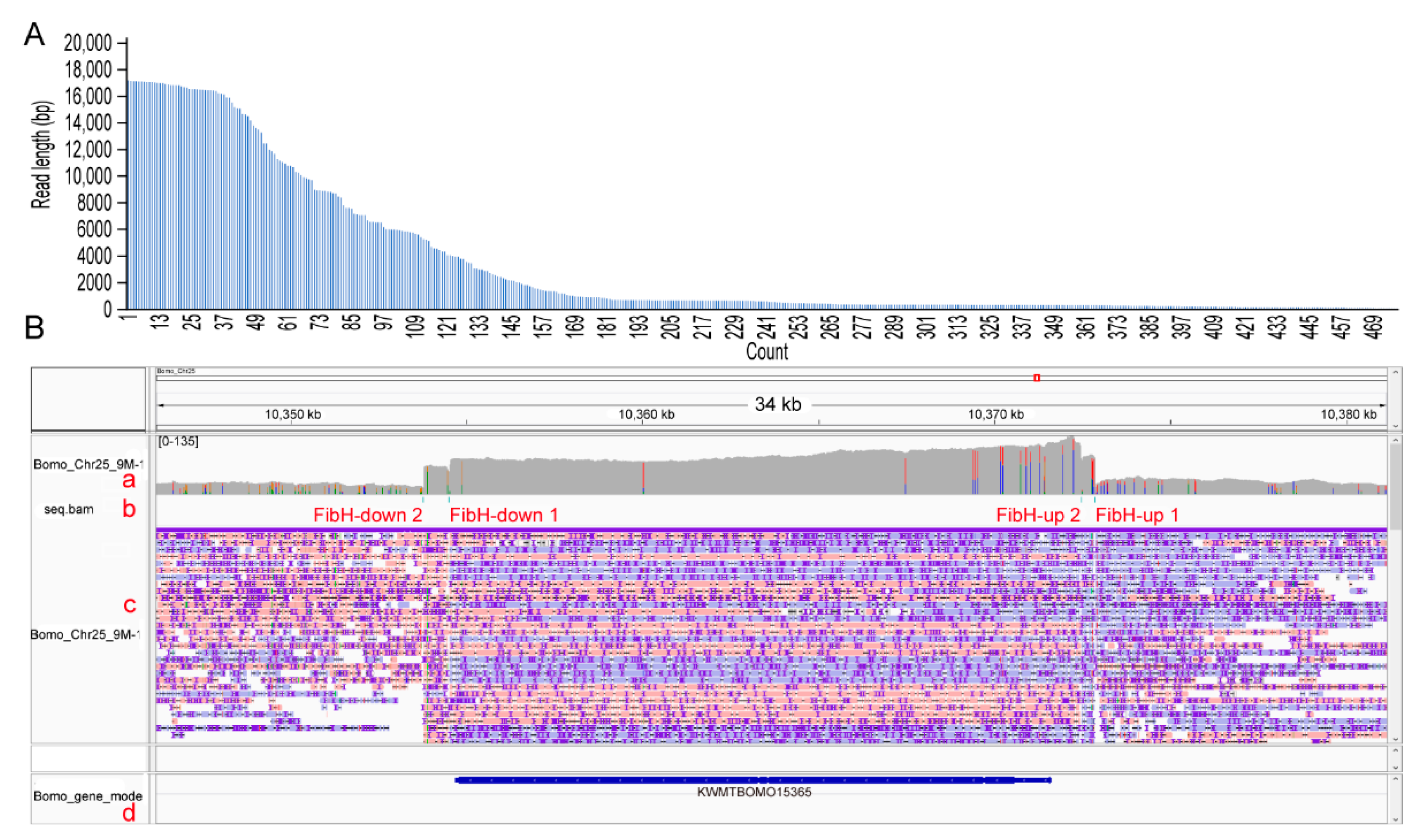
3.3. CEO Could Effectively Enrich the Sequence of Interest
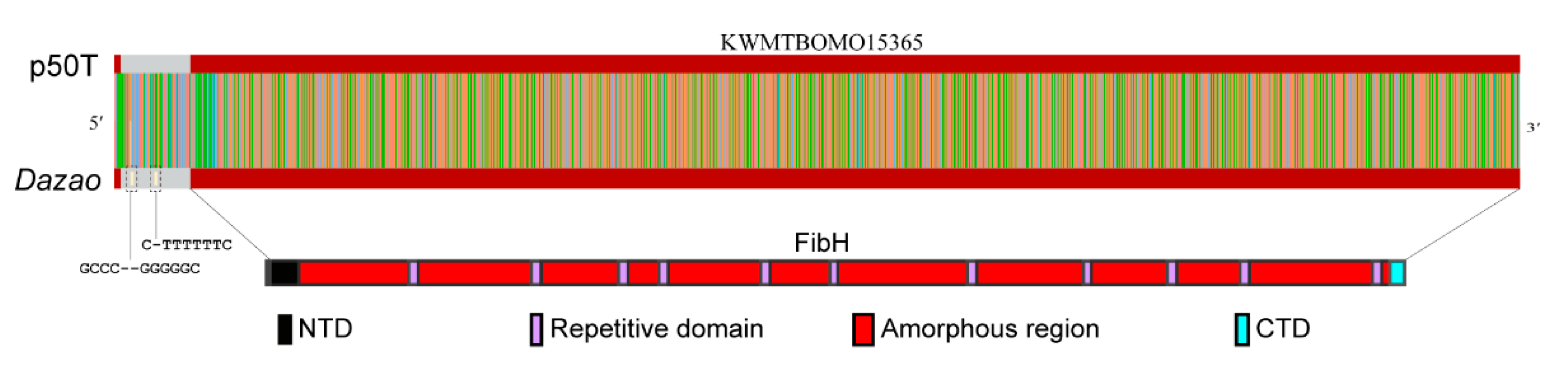
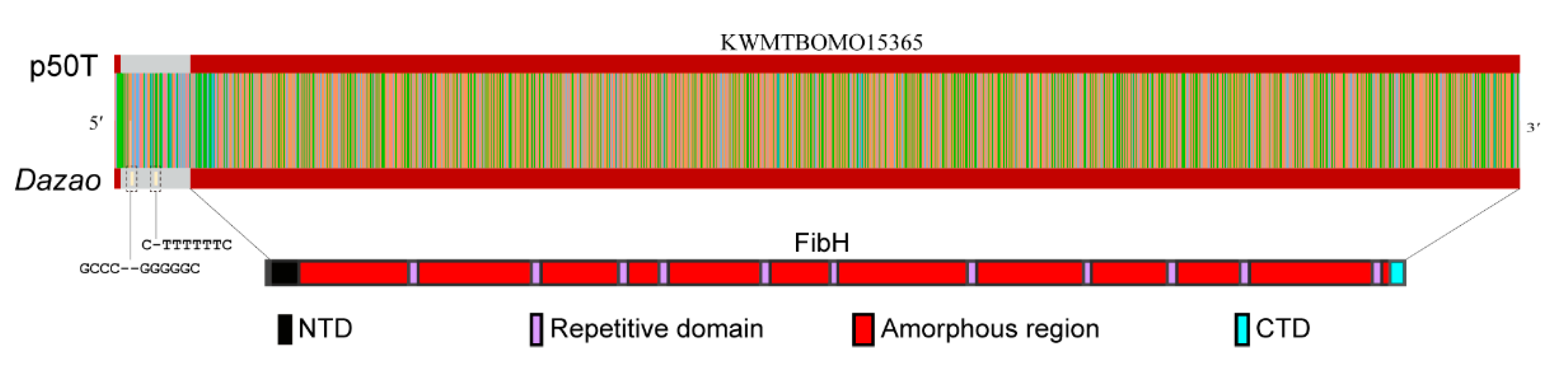
3.4. CEO Could Characterize the Fine Structure of FibH
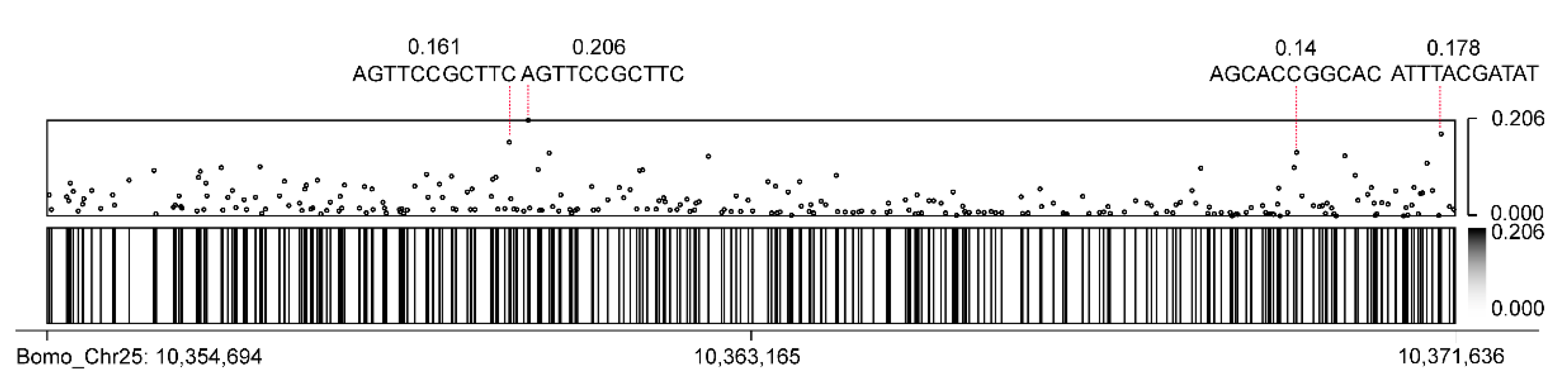
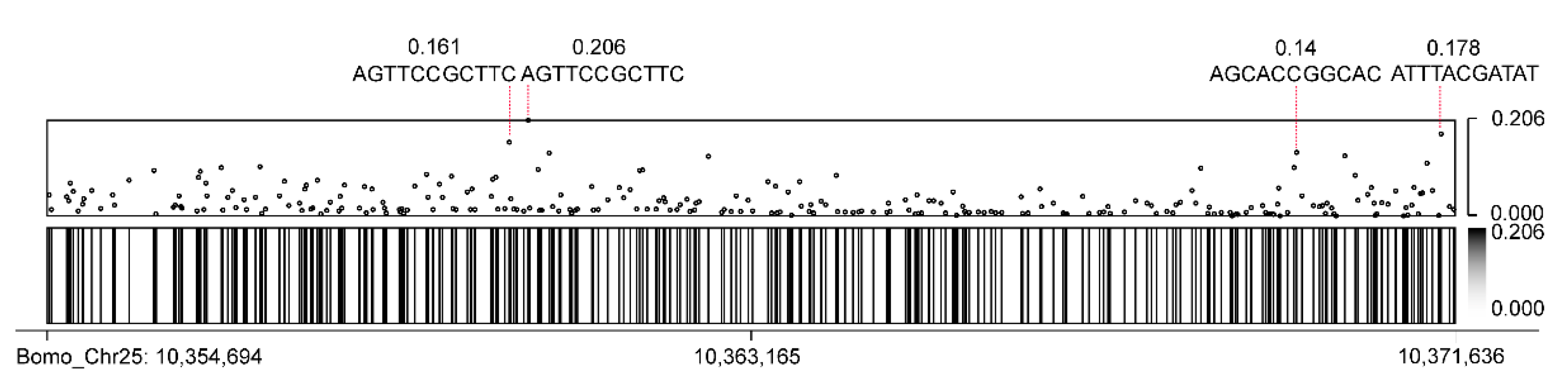
3.5. CEO Could Identify Methylation of FibH
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Panabières, F.; Rancurel, C.; da Rocha, M.; Kuhn, M.-L. Characterization of Two Satellite DNA Families in the Genome of the Oomycete Plant Pathogen Phytophthora parasitica. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Willems, T.; Gymrek, M.; Highnam, G.; Consortium, G.P.; Mittelman, D.; Erlich, Y. The landscape of human STR variation. Genome Res. 2014, 24, 1894–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, M.; Cummings, R.; Yamashita, Y.M. A conserved function for pericentromeric satellite DNA. eLife 2018, 7. [Google Scholar] [CrossRef]
- Hall, L.L.; Byron, M.; Carone, D.M.; Whitfield, T.W.; Pouliot, G.P.; Fischer, A.; Jones, P.; Lawrence, J.B. Demethylated HSATII DNA and HSATII RNA Foci Sequester PRC1 and MeCP2 into Cancer-Specific Nuclear Bodies. Cell Rep. 2017, 18, 2943–2956. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, B.; Walter, K.; Kreher, S.; Kumar, R.; Hummel, M.; Lenze, D.; Köchert, K.; Bouhlel, M.A.; Richter, J.; Soler, E.; et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat. Med. 2010, 16, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.Y.; Shao, W.; Chang, L.; Yin, Y.; Li, T.; Zhang, H.; Hong, Y.; Percharde, M.; Guo, L.; Wu, Z.; et al. Genomic Repeats Categorize Genes with Distinct Functions for Orchestrated Regulation. Cell Rep. 2020, 30, 3296–3311.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Eickbush, D.G.; Speece, I.; Larracuente, A.M. Heterochromatin-dependent transcription of satellite DNAs in the Drosophila melanogaster female germline. eLife 2021, 10. [Google Scholar] [CrossRef]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Xi, J.; Wang, X.; Yue, D.; Dou, T.; Wu, Q.; Lu, J.; Liu, Y.; Yu, W.; Qiao, K.; Lin, J.; et al. 5′ UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain 2021, 144, 601–614. [Google Scholar] [CrossRef]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S.P. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Kono, N.; Nakamura, H.; Mori, M.; Tomita, M.; Arakawa, K. Spidroin profiling of cribellate spiders provides insight into the evolution of spider prey capture strategies. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wen, R.; Jia, Q.; Liu, X.; Xiao, J.; Meng, Q. Analysis of the Full-Length Pyriform Spidroin Gene Sequence. Genes 2019, 10, 425. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Confalonieri, F.; Medina, N.; Zivanovic, Y.; Esnault, C.; Yang, T.; Jacquet, M.; Janin, J.; Duguet, M.; Perasso, R.; et al. Fine organization of Bombyx mori fibroin heavy chain gene. Nucleic Acids Res. 2000, 28, 2413–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, N.; Nakamura, H.; Ohtoshi, R.; Tomita, M.; Numata, K.; Arakawa, K. The bagworm genome reveals a unique fibroin gene that provides high tensile strength. Commun. Biol. 2019, 2, 148. [Google Scholar] [CrossRef]
- Garel, A.; Deleage, G.; Prudhomme, J. Structure and organization of the Bombyx mori sericin 1 gene and of the sericins 1 deduced from the sequence of the Ser 1B cDNA. Insect Biochem. Mol. Biol. 1997, 27, 469–477. [Google Scholar] [CrossRef]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021. [Google Scholar] [CrossRef]
- Dumbovic, G.; Forcales, S.-V.; Perucho, M. Emerging roles of macrosatellite repeats in genome organization and disease development. Epigenetics 2017, 12, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J.; et al. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Jenjaroenpun, P.; de Loose, A.; Alkam, D.; Ussery, D.W.; Nookaew, I.; Leung, Y.-K.; Ho, S.-M.; Day, J.D.; Rodriguez, A. A novel Cas9-targeted long-read assay for simultaneous detection of IDH1/2 mutations and clinically relevant MGMT methylation in fresh biopsies of diffuse glioma. Acta Neuropathol. Commun. 2020, 8, 87. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Yuen, Z.W.-S.; Srivastava, A.; Daniel, R.; McNevin, D.; Jack, C.; Eyras, E. Systematic benchmarking of tools for CpG methylation detection from nanopore sequencing. Nat. Commun. 2021, 12, 3438. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Genova, A.; Buena-Atienza, E.; Ossowski, S.; Sagot, M. Efficient hybrid de novo assembly of human genomes with wengan. Nat. Biotechnol. 2021, 39, 422–430. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Zhang, F.; Koonin, E.V. SnapShot: Class 2 CRISPR-Cas Systems. Cell 2017, 168, 328–328.e1. [Google Scholar] [CrossRef]
- Zetsche, B.; Abudayyeh, O.O.; Gootenberg, J.S.; Scott, D.A.; Zhang, F. A Survey of Genome Editing Activity for 16 Cas12a Orthologs. Keio J. Med. 2020, 69, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safari, F.; Zare, K.; Negahdaripour, M.; Barekati-Mowahed, M.; Ghasemi, Y. CRISPR Cpf1 proteins: Structure, function and implications for genome editing. Cell Biosci. 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef]
- Tang, X.; Lowder, L.G.; Zhang, T.; Malzahn, A.A.; Zheng, X.; Voytas, D.F.; Zhong, Z.; Chen, Y.; Ren, Q.; Li, Q.; et al. A CRISPR-Cpf1 system for efficient genome editing and transcriptional repression in plants. Nat. Plants 2017, 3, 17018. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.-H.; Kim, J.-S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.; Liu, H.; Zhao, Q.; Liu, B.; Wang, L.; Zhang, J.; Zhu, J.; Bao, R.; Luo, Y. Improved CRISPR-Cas12a-assisted one-pot DNA editing method enables seamless DNA editing. Biotechnol. Bioeng. 2019, 116, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Li, S.; Liu, J.; Zheng, X.; Zhao, G.; Wang, J. The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro. Nucleic Acids Res. 2017, 45. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, S.-T.; Ryu, J.; Kang, B.-C.; Kim, J.-S.; Kim, S.-G. CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat. Commun. 2017, 8, 14406. [Google Scholar] [CrossRef] [Green Version]
- Zhong, G.; Wang, H.; Li, Y.; Tran, M.H.; Farzan, M. Cpf1 proteins excise CRISPR RNAs from mRNA transcripts in mammalian cells. Nat. Chem. Biol. 2017, 13, 839–841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Mori, K.; Mizuno, S. Immunological Identification of the Major Disulfide-Linked Light Component of Silk Fibroin. J. Biochem. 1993, 114, 1–4. [Google Scholar] [CrossRef]
- Peng, Z.; Yang, X.; Liu, C.; Dong, Z.; Wang, F.; Wang, X.; Hu, W.; Zhang, X.; Zhao, P.; Xia, Q. Structural and Mechanical Properties of Silk from Different Instars of Bombyx mori. Biomacromolecules 2019, 20, 1203–1216. [Google Scholar] [CrossRef]
- Watson, C.M.; Crinnion, L.A.; Hewitt, S.; Bates, J.; Robinson, R.; Carr, I.M.; Sheridan, E.; Adlard, J.; Bonthron, D.T. Cas9-based enrichment and single-molecule sequencing for precise characterization of genomic duplications. Lab. Investig. 2019, 100, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.; Cohen, D.; Dubois, A.; Martinez, M.G.; Petitjean, K.; Corlu, A.; Testoni, B.; Hernandez-Vargas, H.; Chemin, I. Cas9-targeted nanopore sequencing reveals epigenetic heterogeneity after de novo assembly of native full-length hepatitis B virus genomes. Microb. Genom. 2021, 7. [Google Scholar] [CrossRef]
- McDonald, T.L.; Zhou, W.; Castro, C.P.; Mumm, C.; Switzenberg, J.A.; Mills, R.E.; Boyle, A.P. Cas9 targeted enrichment of mobile elements using nanopore sequencing. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.D.; Sasani, T.A.; Swanier, J.; Gates, B.L.; Greenland, J.; Pedersen, B.S.; Varley, K.E.; Quinlan, A.R. CaBagE: A Cas9-based Background Elimination strategy for targeted, long-read DNA sequencing. PLoS ONE 2021, 16. [Google Scholar] [CrossRef]
- López-Girona, E.; Davy, M.W.; Albert, N.W.; Hilario, E.; Smart, M.E.M.; Kirk, C.; Thomson, S.J.; Chagné, D. CRISPR-Cas9 enrichment and long read sequencing for fine mapping in plants. Plant Methods 2020, 16. [Google Scholar] [CrossRef]
- Hafford-Tear, N.J.; Tsai, Y.-C.; Sadan, A.N.; Sanchez-Pintado, B.; Zarouchlioti, C.; Maher, G.J.; Liskova, P.; Tuft, S.J.; Hardcastle, A.J.; Clark, T.A.; et al. CRISPR/Cas9-targeted enrichment and long-read sequencing of the Fuchs endothelial corneal dystrophy-associated TCF4 triplet repeat. Genet. Med. 2019, 21, 2092–2102. [Google Scholar] [CrossRef]
- Stevens, R.C.; Steele, J.L.; Glover, W.R.; Sanchez-Garcia, J.F.; Simpson, S.D.; O’Rourke, D.; Ramsdell, J.S.; MacManes, M.D.; Thomas, W.K.; Shuber, A.P. A novel CRISPR/Cas9 associated technology for sequence-specific nucleic acid enrichment. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Kawamoto, M.; Jouraku, A.; Toyoda, A.; Yokoi, K.; Minakuchi, Y.; Katsuma, S.; Fujiyama, A.; Kiuchi, T.; Yamamoto, K.; Shimada, T. High-quality genome assembly of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2019, 107, 53–62. [Google Scholar] [CrossRef]
- Stemmer, M.; Thumberger, T.; Del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, T.; Wang, G.; Bi, A.; Wassie, M.; Xie, Y.; Cao, L.; Xu, H.; Fu, J.; Chen, L.; et al. Simultaneous gene editing of three homoeoalleles in self-incompatible allohexaploid grasses. J. Integr. Plant Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Shola, D.T.N.; Yang, C.; Kewaldar, V.-S.; Kar, P.; Bustos, V. New Additions to the CRISPR Toolbox: CRISPR-CLONInG and CRISPR-CLIP for Donor Construction in Genome Editing. CRISPR J. 2020, 3, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Poggi, L.; Emmenegger, L.; Descorps-Declère, S.; Dumas, B.; Richard, G.-F. Differential efficacies of Cas nucleases on microsatellites involved in human disorders and associated off-target mutations. Nucleic Acids Res. 2021, 49, 8120–8134. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef]
- Van Haasteren, J.; Munis, A.M.; Gill, D.R.; Hyde, S.C. Genome-wide integration site detection using Cas9 enriched amplification-free long-range sequencing. Nucleic Acids Res. 2021, 49. [Google Scholar] [CrossRef]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y.; et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Fang, L.; Yu, G.; Wang, D.; Xiao, C.-L.; Wang, K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Gigante, S.; Gouil, Q.; Lucattini, A.; Keniry, A.; Beck, T.; Tinning, M.; Gordon, L.; Woodruff, C.; Speed, T.P.; Blewitt, M.E.; et al. Using long-read sequencing to detect imprinted DNA methylation. Nucleic Acids Res. 2019, 47. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.; Martin-Trujillo, A.; Rodriguez, O.L.; Gies, S.J.; Hadelia, E.; Jadhav, B.; Jain, M.; Paten, B.; Sharp, A.J. Pervasive cis effects of variation in copy number of large tandem repeats on local DNA methylation and gene expression. Am. J. Hum. Genet. 2021, 108, 809–824. [Google Scholar] [CrossRef]
- Stangl, C.; de Blank, S.; Renkens, I.; Westera, L.; Verbeek, T.; Valle-Inclan, J.E.; González, R.C.; Henssen, A.G.; van Roosmalen, M.J.; Stam, R.W.; et al. Partner independent fusion gene detection by multiplexed CRISPR-Cas9 enrichment and long read nanopore sequencing. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, T.; Toyota, T.; Miyatake, S.; Mitsuhashi, S.; Doi, H.; Kudo, Y.; Kishida, H.; Hayashi, N.; Tsuburaya, R.S.; Kinoshita, M.; et al. Complete sequencing of expanded SAMD12 repeats by long-read sequencing and Cas9-mediated enrichment. Brain 2021, 65, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Gabrieli, T.; Sharim, H.; Fridman, D.; Arbib, N.; Michaeli, Y.; Ebenstein, Y. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M.; McClain, M.E.; Martinez, R.A.; Snow, K.; Lipe, H.; Ravits, J.; Bird, T.D.; La Spada, A.R. Late-onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology 2000, 55, 569–572. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| crRNA | Target Sequence * | Genomic Location | GC (%) | Efficiency | MM0 | MM1 | MM2 | MM3 | Size (bp) |
|---|---|---|---|---|---|---|---|---|---|
| FibH-up 1 | TTTATGTTACCGGGGTCTAGTGAC | Chr25: 10,372,894–10,372,871 | 55 | 60 | 0 | 0 | 0 | 0 | 20 |
| FibH-up 2 | TTTAAGCTTGTTGTACAAAACTGC | Chr25: 10,372,500–10,372,477 | 40 | 52 | 0 | 0 | 0 | 1 | 20 |
| FibH-down 1 | TTTATATGAACCTATTGTAATTTAG | Chr25: 10,354,516–10,354,540 | 24 | 59 | 0 | 0 | 0 | 0 | 21 |
| FibH-down 2 | TTTGTACCCTCATACCTCAAAGAAC | Chr25:10,353,778–10,353,802 | 43 | 42 | 0 | 0 | 0 | 0 | 21 |
| Chromosome | Description | Start | End | Average Depth | Enrichment Fold |
|---|---|---|---|---|---|
| Bomo_Chr25 | downstream sequence | 10,353,778 | 10,354,516 | 60.05 | 1.58 |
| Bomo_Chr25 | KWMTBOMO15365 | 10,354,516 | 10,372,477 | 87.07 | 2.29 |
| Bomo_Chr25 | upstream sequence | 10,372,477 | 10,372,871 | 80.12 | 2.11 |
| Motif Name | Motif Sequence | Motif Repetition | Methylation Frequency * |
|---|---|---|---|
| motif-1 | TGCTCCGTATC | 145 | 2.121 |
| motif-2 | AGCACCGGCAC | 92 | 1.782 |
| motif-3 | AGCTCCGCTTC | 65 | 3.251 |
| motif-4 | ATATCCGCCAT | 11 | 0.267 |
| motif-5 | TACTCCGTATC | 10 | 0.222 |
| motif-6 | TGAACCGGCAC | 9 | 0.123 |
| motif-7 | AGCTCCGGCAC | 8 | 0.123 |
| motif-8 | TGCTCCGTACC | 8 | 0.166 |
| motif-9 | AGAACCGGCAC | 3 | 0.092 |
| motif-10 | AGTTCCGCTTC | 3 | 0.438 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, W.; Lan, X.; Zhang, T.; Sun, H.; Ma, S.; Xia, Q. Precise Characterization of Bombyx mori Fibroin Heavy Chain Gene Using Cpf1-Based Enrichment and Oxford Nanopore Technologies. Insects 2021, 12, 832. https://doi.org/10.3390/insects12090832
Lu W, Lan X, Zhang T, Sun H, Ma S, Xia Q. Precise Characterization of Bombyx mori Fibroin Heavy Chain Gene Using Cpf1-Based Enrichment and Oxford Nanopore Technologies. Insects. 2021; 12(9):832. https://doi.org/10.3390/insects12090832
Chicago/Turabian StyleLu, Wei, Xinhui Lan, Tong Zhang, Hao Sun, Sanyuan Ma, and Qingyou Xia. 2021. "Precise Characterization of Bombyx mori Fibroin Heavy Chain Gene Using Cpf1-Based Enrichment and Oxford Nanopore Technologies" Insects 12, no. 9: 832. https://doi.org/10.3390/insects12090832
APA StyleLu, W., Lan, X., Zhang, T., Sun, H., Ma, S., & Xia, Q. (2021). Precise Characterization of Bombyx mori Fibroin Heavy Chain Gene Using Cpf1-Based Enrichment and Oxford Nanopore Technologies. Insects, 12(9), 832. https://doi.org/10.3390/insects12090832